


Em 2001 foi publicada a primeira versão do genoma humano e, finalmente, pudemos, literalmente, ler o DNA presente em humanos. O projeto é considerado por muitos como o de maior proporção na área biológica, tendo por volta de 10 anos de duração, financiamento de três bilhões de dólares e envolvendo pesquisadores do mundo inteiro! Como é de se imaginar, foi uma tarefa nada trivial na época, porém graças a avanços na tecnologia e bioinformática, o projeto foi concluído com uma eficiência maior que a prevista.
Acredito ser de conhecimento geral que muitas de nossas características são passadas de pais para filhos, desde cor dos olhos até predisposição a doenças, como os diversos tipos de câncer. Estas informações estão dentro de genes, cujo conjunto compõe parte de nosso genoma, que está guardado dentro de cada uma de nossas células e são importantíssimas em moldar as nossas vidas. Um exemplo muito interessante envolve a intolerância à lactose. Ela ocorre em pessoas que apresentam pequenas mudanças no gene que codifica a proteína lactase, uma enzima que digere a lactose, fazendo com que o nível da enzima seja menor que o ideal em pessoas afetadas. A ideia do sequenciamento de genomas é justamente ler estas informações e entendermos melhor a biologia de cada organismo.
Desde a década de 80 havia grande interesse em sequenciar o genoma humano mas na época, esta tarefa não parecia nem um pouco viável, afinal estimava-se um genoma do tamanho de 3 bilhões de nucleotídeos, ou seja, 3 bilhões de letras! Mesmo que o conceito de nucleotídeo não esteja claro para você, imagine que na época os maiores genomas já sequenciados eram por volta de 200 mil nucleotídeos, mais de mil vezes menor que o humano. Quando o projeto Genoma Humano começou, em 1990, era claro que a tecnologia de sequenciamento deveria evoluir para tornar o objetivo uma realidade. O fato problemático é que nenhuma máquina de sequenciamento, nem mesmo as atuais, consegue ler o genoma humano de uma vez só.

O grande cientista Craig Venter introduziu uma nova abordagem para este problema. A ideia é criar várias cópias do genoma, fragmentá-las em muitos e muitos fragmentos (por volta de 1500 a 2000 nucleotídeos de tamanho), e finalmente sequenciar cada um destes, gerando os chamados reads. A partir daí, como se fosse um quebra-cabeça, nós utilizamos os reads para montar o genoma original. Já que, anteriormente, diversas cópias do genoma foram geradas, o processo de sequenciamento leu a mesma região genômica mais de uma vez e portanto, há pares de reads que apresentam sobreposição e podemos, computacionalmente, combinar todos os reads e, em uma situação ideal, formar o genoma completo. Em um esforço que durou muitos anos, a primeira versão do genoma foi publicada em 2001 e o projeto dado como concluído em 2003.
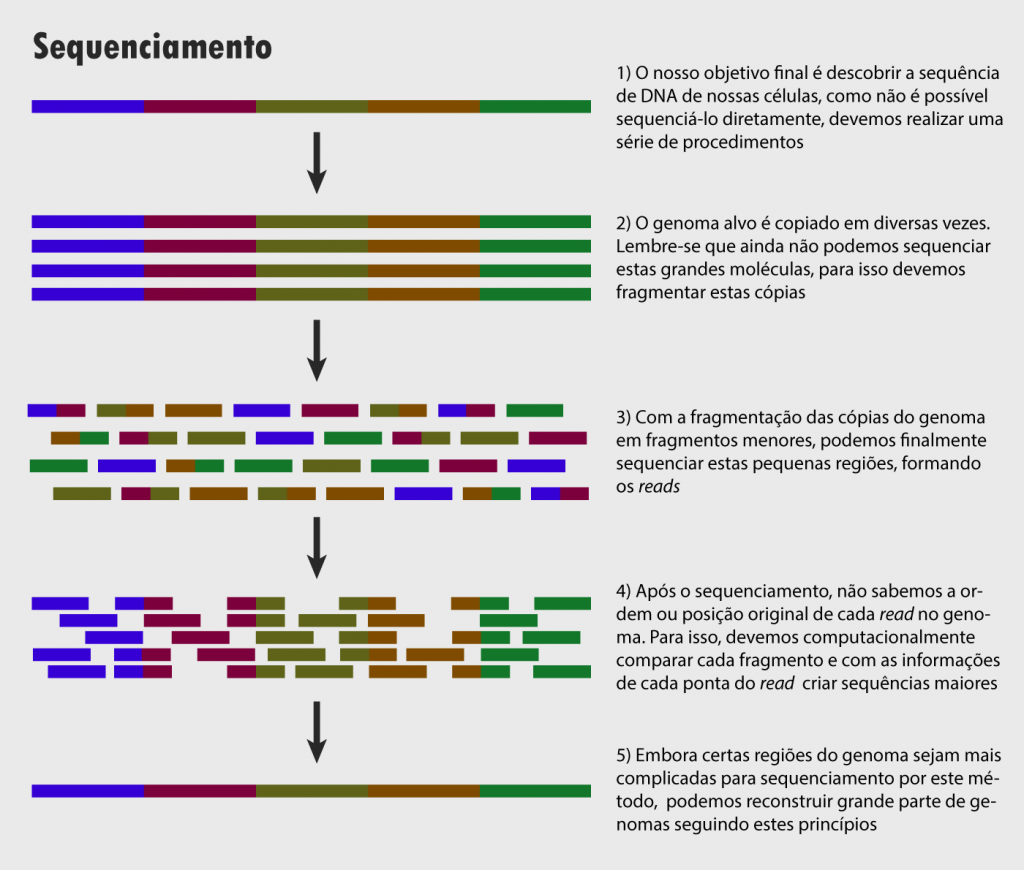
Hoje em dia, já sequenciamos centenas de espécies, e para algumas espécies, muitos indivíduos diferentes (há centenas de genomas humanos já sequenciados). O princípio ainda é o mesmo, porém a escala é muito maior. O custo de sequenciamento por nucleotídeo é 4000 vezes menor do que era ao término do projeto Genoma Humano, mas em troca disso, os reads que geramos são muito menores, em geral entre 100 e 500 nucleotídeos. Somando a quantidade de dados (um sequenciador considerado médio nos fornece dados na magnitude de bilhões de nucleotídeos!) e o pequeno tamanho dos reads, é de se imaginar que não é nada trivial criar um software de montagem que seja rápido, eficiente e acurado o suficiente para a necessidade dos cientistas.
Este projeto é um exemplo claro sobre como a bioinformática tem um aspecto muito dinâmico. Novas técnicas moleculares demandam novas formas de análises, novos algoritmos, novas abordagens para entendermos os dados que estão sendo gerados. E como se fosse um ciclo, os resultados destas novas abordagens agregam ao nosso conhecimento científico e nos permite evoluir e ir além da fronteira do que já é conhecido. Graças à evolução da tecnologia de sequenciamento, potência computacional e algoritmos de montagem podemos, de maneira rotineira, entender o que está dentro de nossas células, o que nos faz ser humanos.