


Como mencionado em uma postagem recente do Blog (https://www.blogs.unicamp.br/tb-of-life/pt_BR/2017/01/22/metagenomica-alem-do-evidente/), a metagenômica consiste em (tentar) identificar todos os micro-organismos de um ambiente. Justamente por querermos identificar os representantes dessa comunidade, à partir de dados de sequenciamento NGS, podemos também obter os genes de cada espécie presente no ambiente de interesse. Essa estratégia para obter o sequenciamento total dos micro-organismos de um determinado ambiente é chamado de Whole Genome Shotgun (WGS).
Antes de mencionarmos e compararmos preditores de genes de metagenômica, utilizando dados reais, discutiremos um pouco sobre o quanto devemos sequenciar para obtermos o maior número de espécies possíveis. Como medir a riqueza e a diversidade das espécies presentes no ambiente? A riqueza e a diversidade de espécies dependem, além da própria natureza da comunidade, do esforço amostral despendido, uma vez que o número de espécies aumenta com o aumento do número de indivíduos amostrados. As curvas de acumulação de espécies permitem avaliar o quanto um estudo se aproxima de capturar todas as espécies do local (Biswas, S. R., and A. U. Mallik. 2011).
A relação entre riqueza e tamanho da amostra não é linear (Schilling and Batista, 2008), mas sabemos que em comunidades com maior uniformidade, o número de espécies total é obtido com um menor tamanho de amostra. Ao ‘graficar’ uma curva que verifique o tamanho da amostra (número de indivíduos) pelo número de espécies acumuladas (riqueza), teremos uma curva como a da Figura 1.
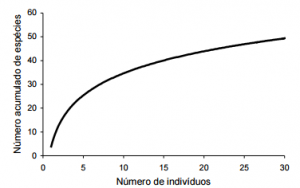
Figura 1. Curva referente a número de indivíduos pelo total de espécies acumuladas sobre uma certa comunidade.
Na Figura 1, podemos notar que a relação entre os eixos não é linear. Quando a curva estabiliza, ou seja, nenhuma espécie nova é adicionada, significa que a riqueza total foi obtida. Nesse caso esquematizado, ao atingir 30 indivíduos, ainda não temos o máximo (patamar, ou plateu) de riqueza obtida. Em todo caso, a estabilização da curva é bastante difícil, pois precisamos adicionar muitas espécies raras para assim atingir o patamar.
A técnica de rarefação, que consiste em calcular o número esperado de espécies em cada amostra para um tamanho de amostra padrão, permite compararmos comunidades e suas riquezas. O número esperado de espécies é obtido pela equação: 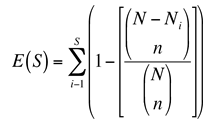
onde E(S) é o número esperado de espécies em uma amostragem aleatória, S é o número total de espécies registradas, N é o número total de indivíduos registrados, Ni é o número de indivíduos da espécie i, e n é o tamanho padronizado da amostra escolhido.
Se colocarmos de forma gráfica, um exemplo de comparação entre duas comunidades seria dado pela Figura 2. A seta vermelha representa o ponto onde as curvas podem ser comparáveis, já que a amostragem se mostrou diferente e rarefação também. NÃO SE DEVE comparar a riqueza total ao comparar comunidades, e sim o ponto de menor riqueza.
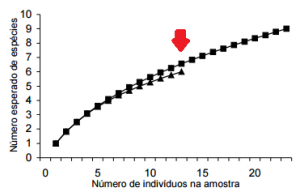 Figura 2. Curva referente a comparação de duas comunidades em relação ao número de indivíduos e total de espécies acumuladas .
Figura 2. Curva referente a comparação de duas comunidades em relação ao número de indivíduos e total de espécies acumuladas .
Extrapolando para a metagenômica, a curva de rarefação se mostra muito importante para estimação de riqueza. O detalhe é que o número de espécies é trocado para o número de OTUs (Operational Taxonomic Unit) e o número de indivíduos é trocado pelo número de reads sequenciados. Essa troca representa uma composição entre o quanto devemos sequenciar para obter o maior número de espécies possíveis. Essa comparação também segue os mesmos passos de não-linearidade como as outras curvas mostradas. Um exemplo de uma curva de rarefação é mostrado na Figura 3. O software utilizado para gerar essas curvas foi o QIIME.
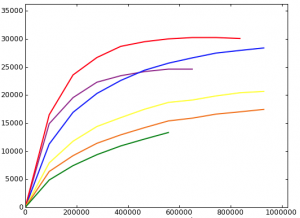
Figura 3. Exemplo de curvas de rarefação. Note que a curva roxa está rarefeita, enquanto a verde ainda não.
Outras medidas de riqueza de espécies possibilitam estimar o número total de espécies numa determinada comunidade a partir dos dados amostrais. Entre estas estão os estimadores Jackknife (1 e 2) e CHAO (1 e 2) (Estudo comparativo dos estimadores [3]). A vantagem desses estimadores é a existência de equações para o cálculo de limites de confiança da estimativa. Esses estimadores surgiram na Ecologia na década de 50 e são utilizados até hoje.
O autor deste post gosta, particularmente, do índice de riqueza CHAO 1. O método Chao 1 estima a riqueza total utilizando o número de espécies representadas por apenas um indivíduo nas amostras (singletons), e o número de espécies com apenas dois indivíduos nas amostras (doubletons). A estimativa de riqueza é calculada pela equação:
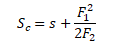
onde Sc é a riqueza estimada, s é a riqueza observada, Fi é o número de espécies que têm exatamente i indivíduos em todas as amostras. Se uma amostra contém muitos singletons, é provável que existam mais OTUs não detectadas, assim, o índice CHAO 1 estimará uma maior riqueza de espécies do que seria para uma amostra sem OTUs raras.
Já que mencionamos sobre curvas e suas peculiaridades, vamos falar sobre os preditores de genes e suas qualidades ou deficiências. Predizer um gene (gene prediction ou gene finding, em inglês) é identificar, através de algoritmos de semelhança de sequência, usualmente sequências de DNA, as que são biologicamente funcionais. Normalmente encontramos genes codificadores de sequência, mas podemos obter genes de RNA ou regiões reguladoras. Decidimos então testar os dois maiores preditores de genes em dados de metagenômica: o Glimmer-MG e o MetaGeneMark.
O algoritmo do MetaGeneMark introduziu os modelos de cadeia de Markov não -homogêneos de periodicidade três, a partir de sequências de DNA, para codificação de proteínas padrões na predição de genes, bem como abordagem Bayesiana para a predição de genes em duas cadeias de DNA, simultaneamente. Os parâmetros específicos das espécies dos modelos são estimados a partir de conjuntos de sequência conhecidas (protein-coding and non-coding). O passo principal do algoritmo calcula, para um dado fragmento de DNA, as probabilidades posteriores de serem “codificadoras de proteínas” (portadoras de código genético) em cada um dos seis quadros de leitura possíveis (incluindo três quadros na cadeia de DNA complementar) ou de serem “não codificantes” (Noguchi,H. et. al, 2006).
O GLIMMER ( Kelley, D.R. et al, 2012) procura principalmente ORFS longas. Usando estas ORFS e seguindo certa distribuição de aminoácidos, o programa gera dados de conjunto de treinamento. Posteriormente, usando esses dados de treinamento, o GLIMMER treina todos os seis modelos de Markov de codificação de DNA e o modelo para DNA não codificante. Um terceiro passo é aplicado, onde o GLIMMER tenta calcular as probabilidades a partir dos dados. Com base no número de observações, ele determina se deve usar modelo de Markov de ordem fixa ou modelo de Markov interpolado. O GLIMMER obtém pontuação para cada ORF longa gerada utilizando todos os seis modelos de DNA codificadores e também usando o modelo de DNA não codificante. Se a pontuação obtida no passo anterior for maior que um determinado limiar, ele prevê que ele seja um gene.
Testamos ambos os preditores de genes para três características finais de análise: tempo de processamento, número de genes preditos e tamanho médio dos genes. Tais características auxiliam na identificação da qualidade da montagem e da predição. O tempo de processamento é razoável para melhorar o tempo de análise; o número de genes e seu tamanho médio referem-se à robustez do algoritmo em predizer genes funcionais longos. Em ambas as análises dos dois preditores foram utilizados os mesmos parâmetros para que não interferissem na análise.
As predições foram realizadas em dados previamente montados pelo montador IDBA_UD e, a partir dos contigs, os genes foram preditos. O total de contigs gerados e os resultados das predições do MetaGeneMark e GLIMMER estão resumidos na Tabela 1.
Tabela 1. Resumo dos processos gerados.

Ao analisarmos a tabela acima, notamos que o tempo de processamento é eficiente para um grande número de contigs (biblioteca E). Além disso notamos que o número de genes encontrados por ambos se diferem em média por 12%, um número que pode estar relacionado com a diferença no algoritmo e na fusão de genes. A grande diferença está no tamanho médio dos genes, que para mim, é a métrica mais importante. O MetaGenemark conseguiu reconstruir um maior número de genes com tamanhos em torno de 600bp, enquanto o GLIMMER conseguiu uma reconstrução em torno de 300bp. QUASE O DOBRO!. Teoricamente o MetaGeneMark teria que perder para o tamanho médio pensando na sua extrapolação por 12% em relação ao GLIMMER, mas o resultado foi o contrário.
Após as análises subsequentes de anotação, decidimos escolher o MetaGeneMark como um melhor preditor de genes em dados de metagenômica do que o GLIMMER, pois apresentou um resultado mais robusto sem perder a qualidade.
Auxílio: Cadeia de Marvok (http://www.ece.drexel.edu/gailr/ECE-S690-503/markov_models.ppt.pdf).
Bibliografia
[1] Biswas, S. R., and A. U. Mallik. 2011. Species diversity and functional diversity relationship varies with disturbance intensity. Ecosphere 2(4):art52. doi:10.1890/ES10-00206.1.
[2] SCHILLING, A.C. and BATISTA, J.L.F. Curva de acumulação de espécies e suficiência amostral em florestas tropicais. Revista Brasil. Bot., V.31, n.1, p.179-187, jan.-mar. 2008.
[3] HORTAL, J., BORGES, P. A. V. and GASPAR, C. (2006), Evaluating the performance of species richness estimators: sensitivity to sample grain size. Journal of Animal Ecology, 75: 274–287. doi:10.1111/j.1365-2656.2006.01048.x.
[4] Noguchi H, Park J, Takagi T. MetaGene: prokaryotic gene finding from environmental genome shotgun sequences. Nucleic Acids Research. 2006;34(19):5623-5630. doi:10.1093/nar/gkl723.
[5] Kelley DR, Liu B, Delcher AL, Pop M, Salzberg SL. Gene prediction with Glimmer for metagenomic sequences augmented by classification and clustering. Nucleic Acids Research. 2012;40(1):e9. doi:10.1093/nar/gkr1067.