


FAQ #8 Como ver se a transformação gênica deu certo?

Já colocamos nossos plasmídeos nos bichinhos, mas ainda não acabou. Essa é a hora de saber se tudo que fizemos até aqui deu certo!
Primeiro, colocamos eles na placa de seleção com antibiótico e só aqueles que realmente incorporaram o plasmídeo vão sobreviver porque ganharam um gene amigo de resistência ao antibiótico. Agora pegamos uma parte das células sobreviventes e fazemos um teste usando a PCR (você lembra dessa técnica, né?).
Como testar com a PCR?! Roubamos os plasmídeos dessas células e tentamos multiplicar o pedaço de DNA que foi grudado nele adicionando primers, nucleotídeos e enzimas sob aquecimento e desaquecimento. Se o nosso plasmídeo modificado tiver sido incorporado pelos micro-organismos haverá a multiplicação do pedacinho e podemos vê-los em uma eletroforese. Caso contrário, nada foi copiado e nada vai aparecer na eletroforese. Então temos o sinal de que alguma coisa não deu certo por aqui e as chances de erros deste teste são bem pequenas.
Depois vamos para um teste melhor ainda, o sequenciamento. Este teste diz exatamente qual é a sequência de pares de base da molécula do plasmídeo e acabam de vez suas dúvidas se a transformação deu certo ou não! O sequenciamento começa parecendo um processo de duplicação normal de DNA. Mas além de cadeias molde, enzimas e nucleotídeos normais, há nucleotídeos especiais sintetizados (ddNTP’s) que possuem duas boas propriedades: emitem luz e interrompem o prologamento da cadeia a partir de onde foram adicionados. Então, imagine uma molécula de DNA que possui um par de base A-T em um determinado comprimento e considere que a base A pertence à cadeia molde e a base T à cadeia complementar. Se esta base T for um nucleotídeo especial temos como identificar “quem é” e “qual sua posição” na cadeia, pois ele emitirá uma cor específica para Timina e o comprimento de sua cadeia está interrompido na posição exata (nº 2 em verde). 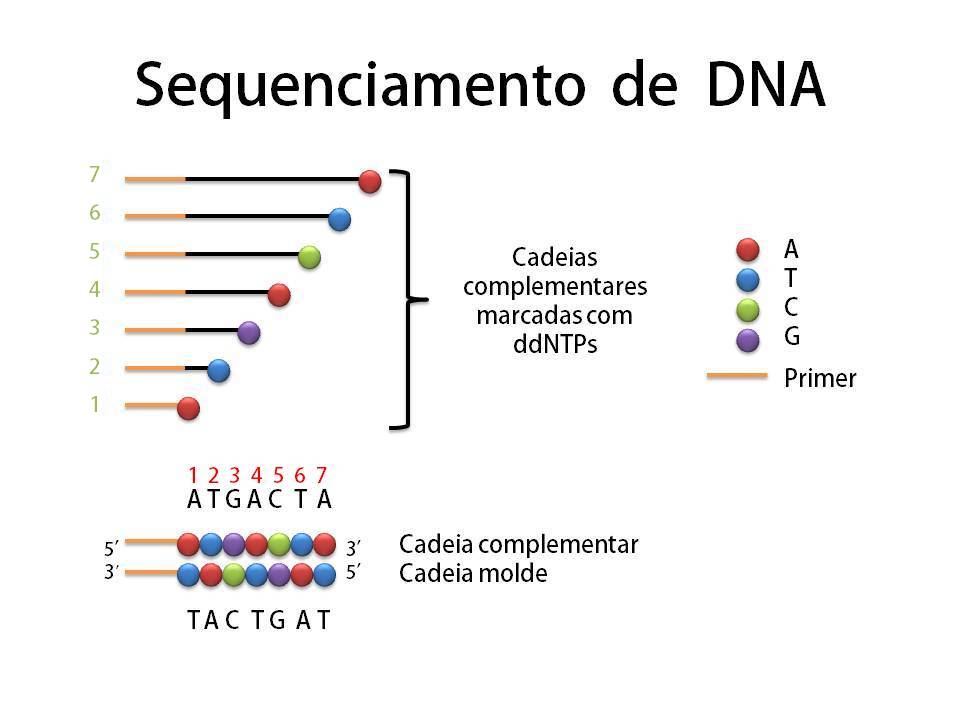 Podemos detectar a cor emitida através de um espectrógrafo e a posição pelo tamanho revelado na eletroforese em gel (o sequenciador é basicamente a união dos dois). Ou detectar o tipo de nucleotídeo presente não pela cor, mas colocando os filamentos “lavados” por cada tipo de nucleotídeo em poços diferentes do gel e interpretando a sequência a olho nu/manualmente. Depois de identificado o nucleotídeo da cadeia complementar é possível saber quem ocupa a mesma posição na cadeia molde, no caso citado é a base Adenina. Expanda esse raciocínio para todas as outras bases da molécula, com uma amostra grande teremos cadeias de todos os comprimentos possíveis, interrompidas por um dos 4 tipos de nucleotídeos especiais (A, T, C, G), e assim é possível sequenciar toda a molécula de DNA!
Podemos detectar a cor emitida através de um espectrógrafo e a posição pelo tamanho revelado na eletroforese em gel (o sequenciador é basicamente a união dos dois). Ou detectar o tipo de nucleotídeo presente não pela cor, mas colocando os filamentos “lavados” por cada tipo de nucleotídeo em poços diferentes do gel e interpretando a sequência a olho nu/manualmente. Depois de identificado o nucleotídeo da cadeia complementar é possível saber quem ocupa a mesma posição na cadeia molde, no caso citado é a base Adenina. Expanda esse raciocínio para todas as outras bases da molécula, com uma amostra grande teremos cadeias de todos os comprimentos possíveis, interrompidas por um dos 4 tipos de nucleotídeos especiais (A, T, C, G), e assim é possível sequenciar toda a molécula de DNA!
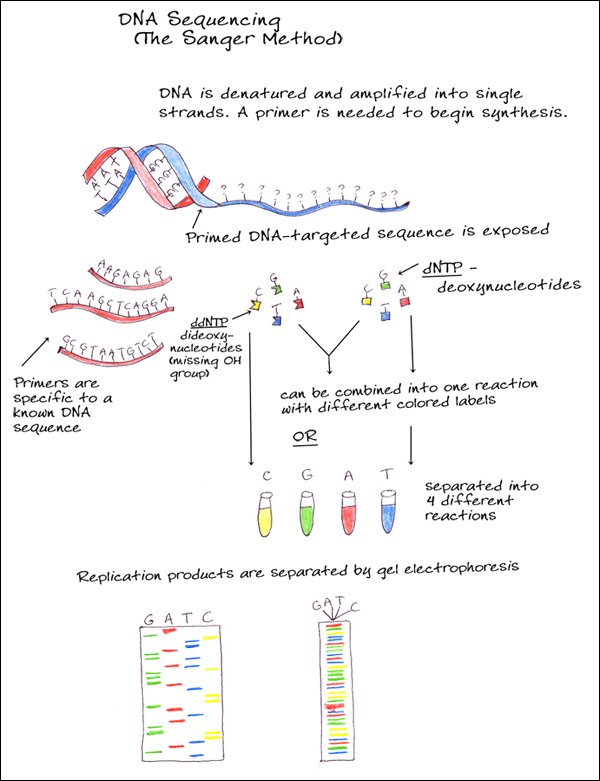
Para saber mais sobre sequenciamento genético, clique aqui.
Por Otto Heringer e Viviane Siratuti.
FAQ #4 Onde colocar esses pedaços de DNA?

“No genoma, né, dêr!”. Certo, mas como? E será que o genoma é o único lugar que podemos colocar esse novo pedacinho de DNA no micro-organismo que queremos modificar? Não! Existe outro lugar também e ele se chama plasmídeo (quem já jogou BioShock vai soltar umas sinapses a mais agora).

O plasmídeo é um DNA circular presente em várias espécies de seres vivos e é responsável por conter informações valiosas envolvendo a sobrevivência do organismo a fatores externos (como por exemplo sua resistência a um antibiótico), além de ser o principal ator da transferência horizontal de informação genética, ou seja, a passagem de um DNA funcional de um ser vivo para outro sem haver hereditariedade (é aí que o jogo BioShock extrapola isso para seres humanos). Adivinha de onde veio a ideia de usar o plasmídeo como transmissor – “vetor” – de informação genética para modificar as células? Veio exatamente desse mecanismo natural de realizar transferência horizontal de genes que vários micro-organismos possuem, então aproveitamos para fazer a transferência das informações genéticas que nós queremos!
E se vamos usar plasmídeos é preciso extraí-los também! Os plasmídeos são moléculas de DNA assim como os pedacinhos obtidos a partir de um genoma, e multiplicados pela PCR, que vamos introduzir no microorganismo, mas aqui há uma etapa importante durante a extração de DNA onde é feita a separação do conteúdo plasmidial do genômico.
O vídeozinho abaixo tem uma animação no ínicio e depois mostra o procedimento do isolamento do plasmídeo em lab!
[youtube_sc url=”http://www.youtube.com/watch?v=8xEDEJ0DHFA”]
Por Otto Heringer e Viviane Siratuti.
Cientistas descobrem novas ferramentas para reescrever o código da vida
O poder de editar genes é revolucionário, útil e com potenciais ilimitados. Porém, a maior parte das ferramentas de edição de DNA são lentas, caras e difíceis de usar – é uma brilhante tecnologia na sua infância. Agora, pesquisadores de Harvard desenvolveram uma técnica que pode editar genomas de uma forma rápida e fácil, reescrevendo o genoma de células vivas. A técnica funciona como um processador de textos, que tem as funções de localizar e substituir. Ele reconhece uma seqüência específica no DNA e a substitui por outra.
“Pela primeira vez, estamos demonstrando que é possível fazer mudanças genômicas no nível do códon”, disse Farren Isaacs, um bioengenheiro da Universidade de Yale em New Haven, Connecticut. “Nós seremos capazes de introduzir novas funcionalidades em organismos”.
A técnica, publicada na revista Science, explora a redundância do código genético. Os aminoácidos, que compõem as proteínas, são codificados por combinações de três letras de DNA chamadas códons. Múltiplos códons às vezes codificam o mesmo aminoácido, por isso se diz que o código genético é degenerado ou redundante.
Isaacs e seus colegas escolheram um códon de parada, TAG, que, junto com o TAA e TGA, sinalizam o fim de uma cadeia de aminoácidos e a liberação da proteína formada. Como esses três códons apresentam a mesma função, os pesquisadores decidiram apagar todos os TAGs do
genoma de uma Escherichia coli e substituí-los por TAAs, utilizando uma plataforma chamada “multiplex automated genome engineering, ou MAGE”. Isso deixa o TAG livre para codificar um novo aminoácido.
Foram sintetizadas 314 fitas de DNA idênticas ao genoma da E. coli exceto que todos os TAGs, nas 314 fitas ao todo, estavam substituídos por TAAs. Em outras palavras, cada uma das 314 fitas não tinha todos os TAGs substituídos por TAAs, mas as 314 fitas juntas sim! Eles então aplicaram corrente elétrica para permitir a entrada do novo DNA nas células. Muitas repetições desta técnica resultaram na obtenção de 31 linhagens da bactéria com 10 dos genes modificados e uma com 4. A equipe bolou um esquema para canalizar todas as 314 mutações para uma célula. Os pesquisadores fizeram uso da habilidade que as bactérias têm de transferir genes para outras bactérias, a conjugação: as 32 linhagens foram pareadas, sendo que uma linhagem doou seus genes mutados para a outra. As 16 linhagens resultantes foram pareadas para formar 8, e novamente para formar 4, condensando as mutações ao longo desse processo. O processo foi batizado “conjugative assembly genome engineering (CAGE)”.
Ansiosos para compartilhar sua tecnologia, eles publicaram seus resultados assim que o CAGE atingiu a rodada semifinal. Os resultados sugerem que as quatro linhagens finais são saudáveis, mesmo com a quantidade de mudanças a que as células foram submetidas.
Após mais duas rodadas de CAGE, segundo Isaacs, uma única linhagem da bactéria conterá todas as 314 mutações e será livre de TAG, que ficará disponível para codificar um aminoácido artificial. Isso desafia as pessoas a imaginarem o genoma como algo muito maleável e editável. Alguns laboratórios já criaram esses aminoácidos, assim como a maquinaria necessária para incorporá-los em proteínas. “O grande avanço aqui é que nós teremos um hospedeiro que permitirá a incorporação de aminoácidos artificiais a taxas muito superiores”, disse Isaacs.
Estes organismos engenheirados seriam geneticamente isolados de outros organismos. A nova informação genética não seria capaz de contaminar organismos naturais porque, fora do laboratório, os aminoácidos naturais no lugar dos artificiais criariam proteínas não funcionais. Além disso, esses organismos seriam imunes a vírus que se baseiam na tradução protéica tradicional – importante para manter linhagens saudáveis e úteis industrialmente. A importância da imunidade viral reside no fato de que indústrias nas quais são cultivadas bactérias, como as farmacêuticas e energéticas, esses vírus podem afetar até 20% das culturas. Um exemplo notável acometeu a Genzyme, cujas perdas devido a contaminações virais podem ter variado de milhões de dólares a até $1 bilhão.
“Essa técnica é mais barata que tentar elaborar genomas a partir do zero. Ao modificar genomas existentes, a maior parte do trabalho já está feita”, disse o co-autor George Church, um geneticista da Harvard Medical School em Boston, Massachusetts.
Cientistas do J. Craig Venter Institute (JCVI), que no ano passado “criaram” a primeira bactéria controlada por um genoma sintético, dizem que o método traz coisas importantes para esse campo de estudo, mas só funciona na prática se o genoma desejado é similar a um organismo existente. “Ultimamente, no JCVI nós estamos tentando fazer células a partir do zero, e apenas uma síntese genômica de novo tornaria isso possível”, disse um porta-voz do Instituto via email.
As duas técnicas provavelmente serão usadas em conjunto, disse Isaacs. “Não surpreenderia se essas tecnologias se unissem e nós começássemos a ver técnicas híbridas inclusive mais poderosas do que as que vemos hoje”.
Referências:
Precise Manipulation of Chromosomes in Vivo Enables Genome-Wide Codon Replacement Isaacs, et al. Science 15 July 2011: 333 (6040), 348-353. [DOI:10.1126/science.1205822]
http://www.eurekalert.org/pub_releases/2011-07/hms-etg071111.php
http://www.nature.com/news/2011/110714/full/news.2011.419.html
[twitter-follow screen_name=’MnlimasBio’]