


FAQ #8 Como ver se a transformação gênica deu certo?

Já colocamos nossos plasmídeos nos bichinhos, mas ainda não acabou. Essa é a hora de saber se tudo que fizemos até aqui deu certo!
Primeiro, colocamos eles na placa de seleção com antibiótico e só aqueles que realmente incorporaram o plasmídeo vão sobreviver porque ganharam um gene amigo de resistência ao antibiótico. Agora pegamos uma parte das células sobreviventes e fazemos um teste usando a PCR (você lembra dessa técnica, né?).
Como testar com a PCR?! Roubamos os plasmídeos dessas células e tentamos multiplicar o pedaço de DNA que foi grudado nele adicionando primers, nucleotídeos e enzimas sob aquecimento e desaquecimento. Se o nosso plasmídeo modificado tiver sido incorporado pelos micro-organismos haverá a multiplicação do pedacinho e podemos vê-los em uma eletroforese. Caso contrário, nada foi copiado e nada vai aparecer na eletroforese. Então temos o sinal de que alguma coisa não deu certo por aqui e as chances de erros deste teste são bem pequenas.
Depois vamos para um teste melhor ainda, o sequenciamento. Este teste diz exatamente qual é a sequência de pares de base da molécula do plasmídeo e acabam de vez suas dúvidas se a transformação deu certo ou não! O sequenciamento começa parecendo um processo de duplicação normal de DNA. Mas além de cadeias molde, enzimas e nucleotídeos normais, há nucleotídeos especiais sintetizados (ddNTP’s) que possuem duas boas propriedades: emitem luz e interrompem o prologamento da cadeia a partir de onde foram adicionados. Então, imagine uma molécula de DNA que possui um par de base A-T em um determinado comprimento e considere que a base A pertence à cadeia molde e a base T à cadeia complementar. Se esta base T for um nucleotídeo especial temos como identificar “quem é” e “qual sua posição” na cadeia, pois ele emitirá uma cor específica para Timina e o comprimento de sua cadeia está interrompido na posição exata (nº 2 em verde). 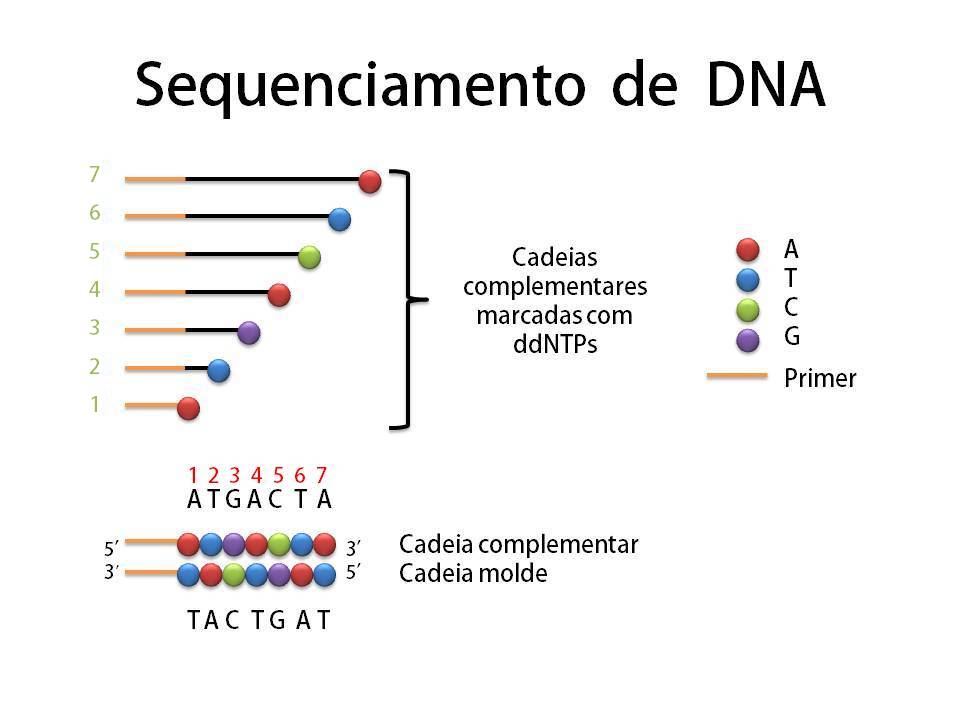 Podemos detectar a cor emitida através de um espectrógrafo e a posição pelo tamanho revelado na eletroforese em gel (o sequenciador é basicamente a união dos dois). Ou detectar o tipo de nucleotídeo presente não pela cor, mas colocando os filamentos “lavados” por cada tipo de nucleotídeo em poços diferentes do gel e interpretando a sequência a olho nu/manualmente. Depois de identificado o nucleotídeo da cadeia complementar é possível saber quem ocupa a mesma posição na cadeia molde, no caso citado é a base Adenina. Expanda esse raciocínio para todas as outras bases da molécula, com uma amostra grande teremos cadeias de todos os comprimentos possíveis, interrompidas por um dos 4 tipos de nucleotídeos especiais (A, T, C, G), e assim é possível sequenciar toda a molécula de DNA!
Podemos detectar a cor emitida através de um espectrógrafo e a posição pelo tamanho revelado na eletroforese em gel (o sequenciador é basicamente a união dos dois). Ou detectar o tipo de nucleotídeo presente não pela cor, mas colocando os filamentos “lavados” por cada tipo de nucleotídeo em poços diferentes do gel e interpretando a sequência a olho nu/manualmente. Depois de identificado o nucleotídeo da cadeia complementar é possível saber quem ocupa a mesma posição na cadeia molde, no caso citado é a base Adenina. Expanda esse raciocínio para todas as outras bases da molécula, com uma amostra grande teremos cadeias de todos os comprimentos possíveis, interrompidas por um dos 4 tipos de nucleotídeos especiais (A, T, C, G), e assim é possível sequenciar toda a molécula de DNA!
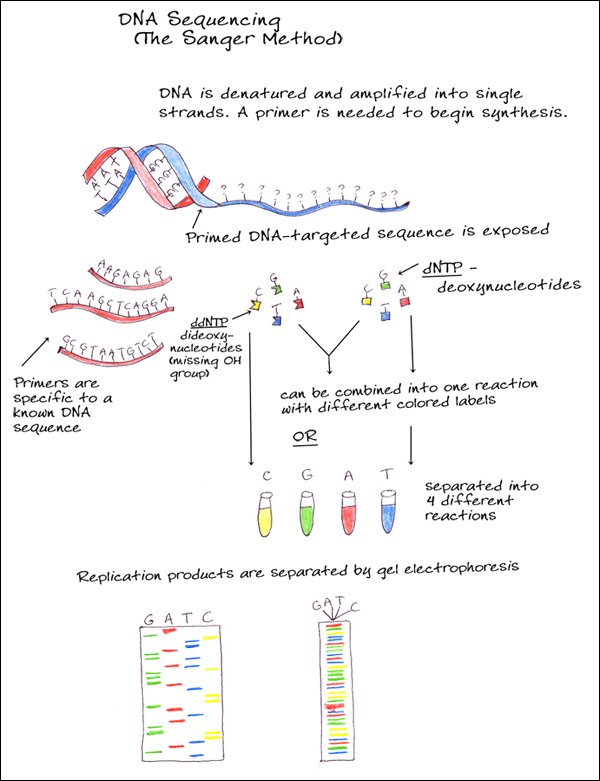
Para saber mais sobre sequenciamento genético, clique aqui.
Por Otto Heringer e Viviane Siratuti.
FAQ #7 Como enfiar o plasmídeo nas células?

Dar e receber plasmídeos não é nenhuma novidade para algumas células!
As bactérias costumam trocar informações genéticas assim e esse processo é chamado de conjugação, portanto elas já possuem toda maquinaria necessária pra isso! Desse jeito elas aumentam muito a variabilidade e as chances de sobrevivência da população.
Podemos usar esse mecanismo natural para enfiar os plasmídeos, mas na maioria das vezes damos uma ajudinha usando choques térmicos ou elétricos.
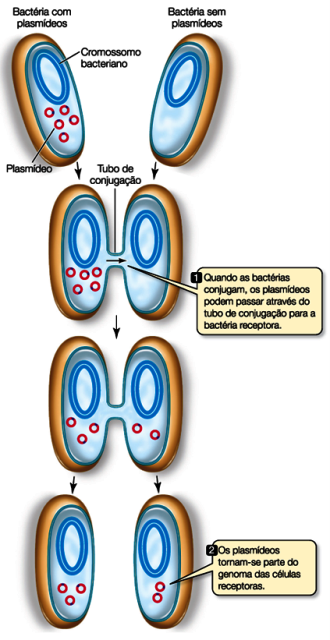
Para entender o choque térmico, assista o vídeo do JOVE aqui. E do choque elétrico, aqui.
Por Otto Heringer e Viviane Siratuti.
FAQ #6 Como ver se nossos plasmídeos incorporaram os pedacinhos de DNA?

Sabendo o tamanho do plasmídeo e dos pedacinhos estimamos um tamanho para nosso novo plasmídeo e agora é só conferir se estão como esperado!
Para isso, existe uma técnica de separação chamada eletroforese em gel, onde as moléculas são “peneiradas” por algum polímero (gel de poliacrilamida ou de agarose) quando se movimentam atraídas ou repulsadas pelos eletrodos de cargas opostas que são colocados nas extremidades do gel. Ou seja, uma molécula com carga negativa caminha para o eletrodo de carga positiva e vice-versa. E as moléculas de mesma carga correm de maneiras diferentes pelo gel de acordo com seus pesos moleculares e tamanhos, as menores e mais “leves” passam com mais facilidade pelo gel enquanto as maiores e mais “pesadas” ficam mais retidas. No final, temos as moléculas separadas ao longo do gel e podemos comparar com a ladder (uma “régua” feita de moléculas com pesos e tamanhos já conhecidos) para saber se nossos plasmídeos estão no tamanho esperado.
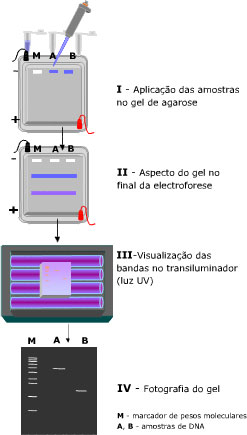
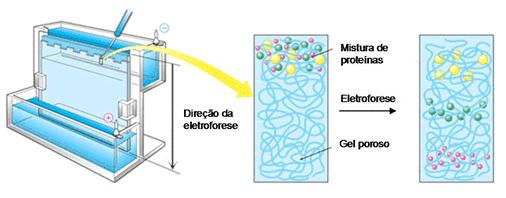
Depois de confirmar pela eletroforese que nossos plasmídeos foram modificados, precisamos “salvá-los” do gel para colocar nas células. Basicamente, cortamos o pedaço de gel que está com nossos plasmídeos e colocamos em tubinhos com uma pequena coluna de sílica dentro (imagem aí embaixo). Então as moléculas de DNA se ligam à coluna, fazemos uma lavagem para tirar o gel e depois diluímos o DNA para retirá-lo da coluna!

Para saber mais sobre eletroforese, assista o vídeo do JOVE Science aqui e purificação aqui. 🙂
Por Otto Heringer e Viviane Siratuti.
FAQ #5 Como colocar os pedaços de DNA no plasmídeo?

Para abrir os plasmídeos (lembrando que é um DNA circular) e depois grudar os pedacinhos de DNA que extraímos pela Miniprep e multiplicamos pela PCR, usamos as chamadas enzimas de restrição e de ligação. Isso tudo ainda fora das células, ok?!
As enzimas de restrição reconhecem e cortam os plasmídeos em regiões específicas que queremos, isto é, cortam em determinadas sequências de pares de base. Inclusive existem dois tipos de corte, podendo formar extremidades coesivas (cortes em comprimentos diferentes entre as duas fitas) ou cegas (corte reto na dupla fita).
Feito isso, agora usamos as enzimas de ligação para grudar as extremidades dos plasmídeos às extremidades dos pedacinhos, onde as bases se complementam.
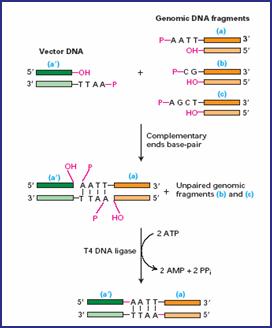
Legal saber também que o nome dessas enzimas estão sempre relacionados com os nomes dos organismos onde elas foram encontradas (veja a enzima EcoRI, por exemplo).

Para assistir o vídeo do JOVE Science sobre enzimas de restrição, clique aqui. E enzimas de ligação, aqui.
Por Otto Heringer e Viviane Siratuti.
FAQs da Bioengenharia – Introdução

Preocupados com os novos amantes da biologia sintética, estamos procurando materiais de introdução no assunto e encontramos algumas coisas interessantes! 🙂
Por isso vamos colocar uma sequência de posts explicativos, partindo de uma visão geral e seguindo por perguntas mais específicas. É um pequeno tutorial-FAQ sobre alguns fundamentos que quando entendidos ajudam bastante a entender algumas de nossas discussões e outros posts deste blog. Serve para ajudar principalmente quem não é da área de biológicas ou quem ainda não se aprofundou muito neste assunto! Se continuarem com dúvidas, perguntem tá?!
Lembrem-se de ativar as legendas dos vídeos, caso necessitem! Todos possuem legendas originais em inglês e alguns em português. Você pode colocá-las traduzidas quando só houver em inglês (não muito recomendado, rs). Nos vídeos do JOVE existe a opção de colocar os textos em português. Se você não sabia que o YouTube fazia isso por você, dá uma olhada aqui (dica: barrinha inferior do vídeo).
Antes de mais nada, você sabe o que são e quais as relações entre nossas principais moléculas orgânicas (DNA, RNA e proteínas), certo? Se sua resposta é não, assista um desses vídeos!
[youtube_sc url=”http://www.youtube.com/watch?v=h3b9ArupXZg#t=0″]
[youtube_sc url=”http://www.youtube.com/watch?v=tMr9XH64rtM”]
Indo além da Estrutura
OK, agora vamos além desses conceitos básicos dos vídeos. Mais que entender a estrutura e a dinâmica dessas biomóleculas, como os cientistas a alteram? Em linhas gerais, como se faz para modificar geneticamente um organismo?
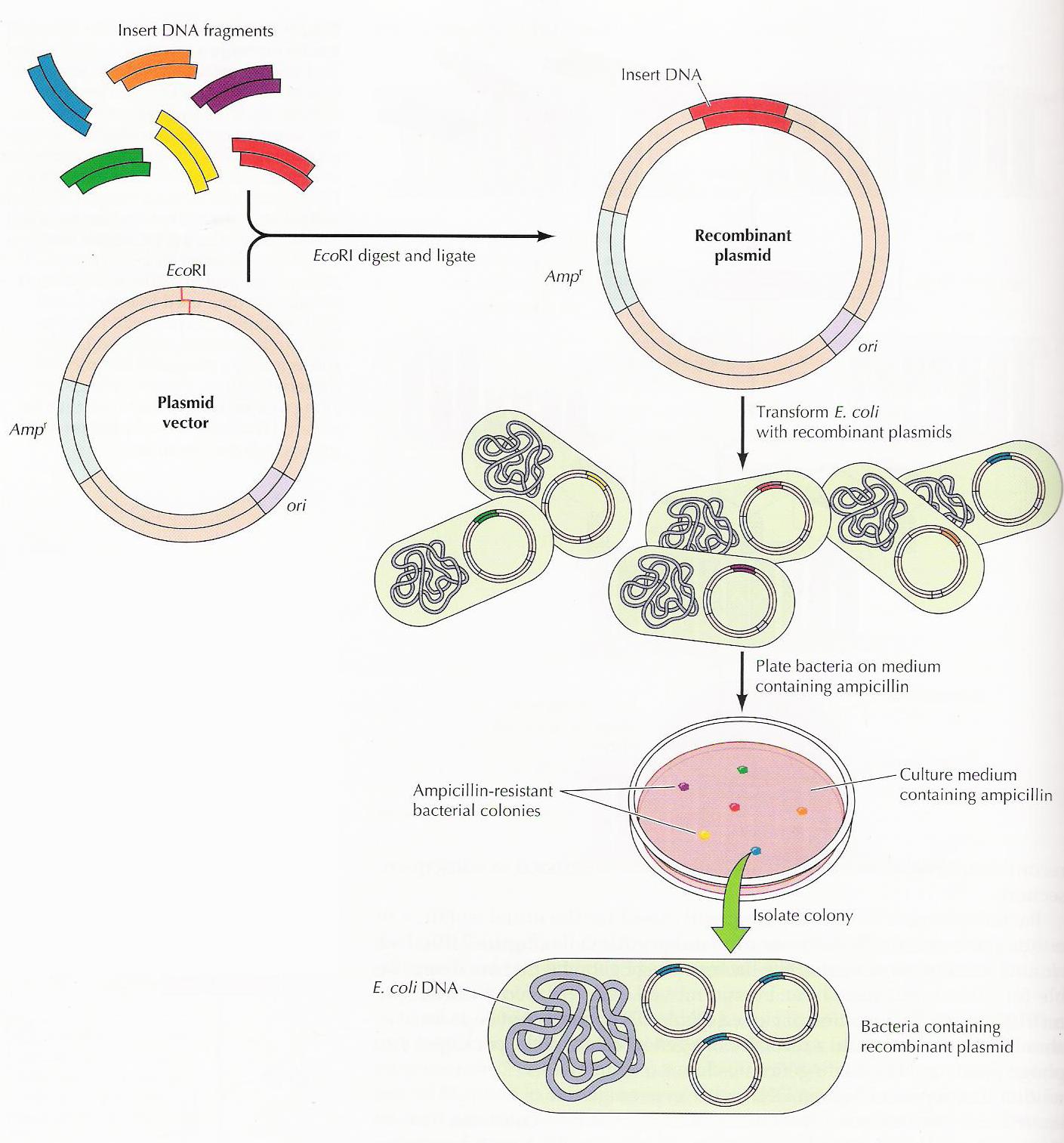
Vamos lá! Primeiro pegamos um pedacinho codificante de DNA (gene) de outro organismo que tenha certa característica que desejamos. Para isso, extraímos o DNA e depois multiplicamos só esse pedaço que nos interessa através de uma técnica chamada PCR (afinal, a eficiência das transformações é pequena e quanto mais material melhor). Precisamos também extrair e abrir os plasmídeos (DNA circular) do organismo escolhido para receber este gene e assim receber as novas características (já que o plasmídeo é nosso principal veículo de introdução dos genes nas células). Para abrir estes plasmídeos usamos enzimas de restrição que cortam as moléculas de DNA em lugares específicos e chamamos isso de digestão. Depois que tivermos vários plasmídeos e genes, podemos grudá-los com a ajuda de enzimas de ligação. E então, para saber se nosso novo plasmídeo deu certo usamos uma técnica de separação por carga e tamanho de molécula, a eletroforese em gel. Nela, as moléculas se acumulam em certas posições que nos dizem seus tamanhos (mergulhadas em um polímero) e assim podemos identificar aquelas que correspondem ao que estimamos. Além disso, fazemos testes com a técnica PCR e o sequenciamento genético. Se os testes disserem que tudo que fizemos deu certo, recuperamos o material e colocamos nossos novos plasmídeos nos organismos, geralmente utilizando choque térmico ou elétrico. Esse passo, introdução de plasmídeos, é a chamada transformação. Mas para selecionarmos somente aqueles organismos que realmente foram transformados usamos antibióticos que matam as células que não ganharam os nossos novos plasmídeos, já que não possuem os genes de resistência que foram colocados neles. E no final, cada célula que sobreviveu se multiplicará, formando colônias de organismos geneticamente modificados. Agora dá uma olhada no esquema de novo e vê se entendeu até aqui!
Por Otto Heringer e Viviane Siratuti.