


FAQ #8 Como ver se a transformação gênica deu certo?

Já colocamos nossos plasmídeos nos bichinhos, mas ainda não acabou. Essa é a hora de saber se tudo que fizemos até aqui deu certo!
Primeiro, colocamos eles na placa de seleção com antibiótico e só aqueles que realmente incorporaram o plasmídeo vão sobreviver porque ganharam um gene amigo de resistência ao antibiótico. Agora pegamos uma parte das células sobreviventes e fazemos um teste usando a PCR (você lembra dessa técnica, né?).
Como testar com a PCR?! Roubamos os plasmídeos dessas células e tentamos multiplicar o pedaço de DNA que foi grudado nele adicionando primers, nucleotídeos e enzimas sob aquecimento e desaquecimento. Se o nosso plasmídeo modificado tiver sido incorporado pelos micro-organismos haverá a multiplicação do pedacinho e podemos vê-los em uma eletroforese. Caso contrário, nada foi copiado e nada vai aparecer na eletroforese. Então temos o sinal de que alguma coisa não deu certo por aqui e as chances de erros deste teste são bem pequenas.
Depois vamos para um teste melhor ainda, o sequenciamento. Este teste diz exatamente qual é a sequência de pares de base da molécula do plasmídeo e acabam de vez suas dúvidas se a transformação deu certo ou não! O sequenciamento começa parecendo um processo de duplicação normal de DNA. Mas além de cadeias molde, enzimas e nucleotídeos normais, há nucleotídeos especiais sintetizados (ddNTP’s) que possuem duas boas propriedades: emitem luz e interrompem o prologamento da cadeia a partir de onde foram adicionados. Então, imagine uma molécula de DNA que possui um par de base A-T em um determinado comprimento e considere que a base A pertence à cadeia molde e a base T à cadeia complementar. Se esta base T for um nucleotídeo especial temos como identificar “quem é” e “qual sua posição” na cadeia, pois ele emitirá uma cor específica para Timina e o comprimento de sua cadeia está interrompido na posição exata (nº 2 em verde). 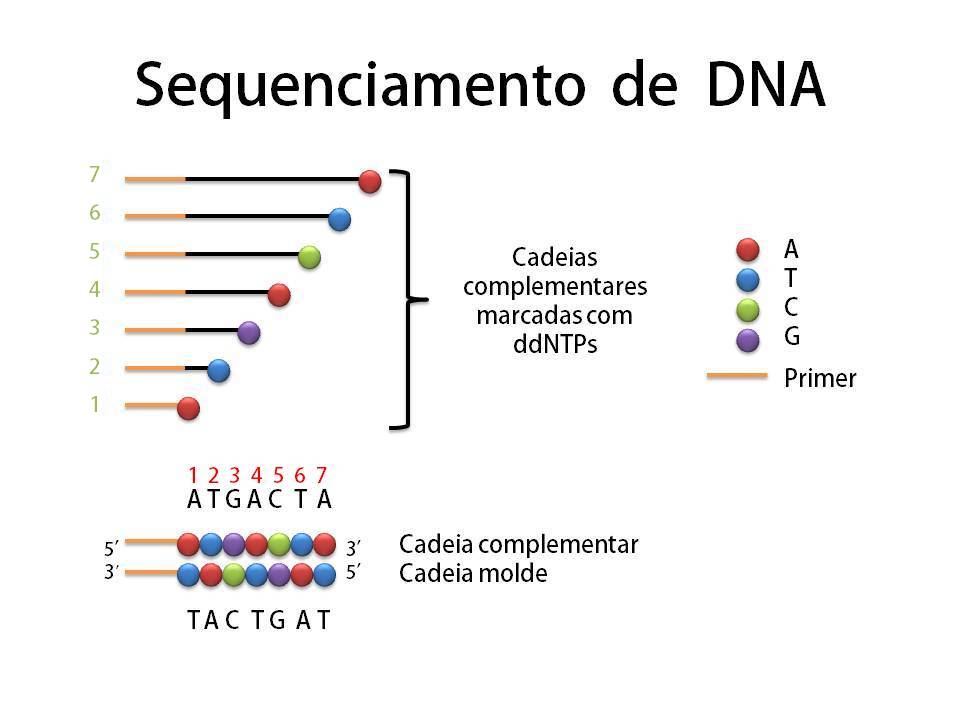 Podemos detectar a cor emitida através de um espectrógrafo e a posição pelo tamanho revelado na eletroforese em gel (o sequenciador é basicamente a união dos dois). Ou detectar o tipo de nucleotídeo presente não pela cor, mas colocando os filamentos “lavados” por cada tipo de nucleotídeo em poços diferentes do gel e interpretando a sequência a olho nu/manualmente. Depois de identificado o nucleotídeo da cadeia complementar é possível saber quem ocupa a mesma posição na cadeia molde, no caso citado é a base Adenina. Expanda esse raciocínio para todas as outras bases da molécula, com uma amostra grande teremos cadeias de todos os comprimentos possíveis, interrompidas por um dos 4 tipos de nucleotídeos especiais (A, T, C, G), e assim é possível sequenciar toda a molécula de DNA!
Podemos detectar a cor emitida através de um espectrógrafo e a posição pelo tamanho revelado na eletroforese em gel (o sequenciador é basicamente a união dos dois). Ou detectar o tipo de nucleotídeo presente não pela cor, mas colocando os filamentos “lavados” por cada tipo de nucleotídeo em poços diferentes do gel e interpretando a sequência a olho nu/manualmente. Depois de identificado o nucleotídeo da cadeia complementar é possível saber quem ocupa a mesma posição na cadeia molde, no caso citado é a base Adenina. Expanda esse raciocínio para todas as outras bases da molécula, com uma amostra grande teremos cadeias de todos os comprimentos possíveis, interrompidas por um dos 4 tipos de nucleotídeos especiais (A, T, C, G), e assim é possível sequenciar toda a molécula de DNA!
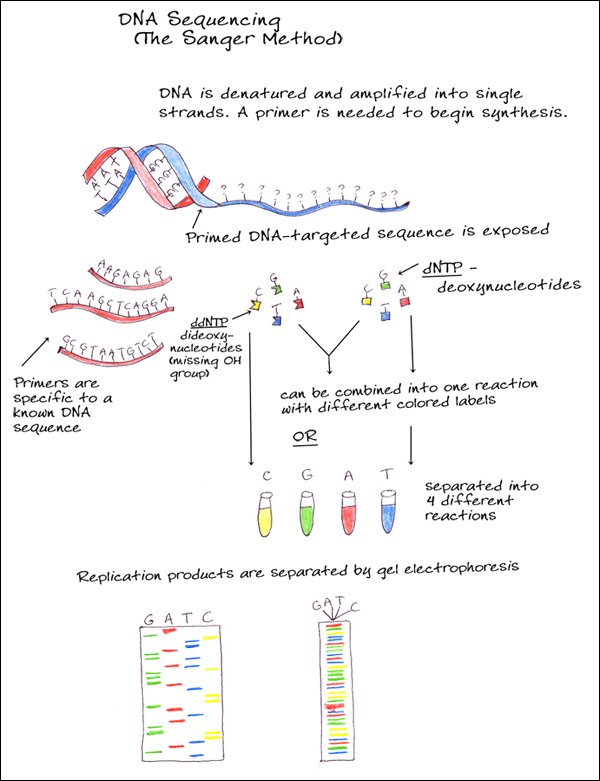
Para saber mais sobre sequenciamento genético, clique aqui.
Por Otto Heringer e Viviane Siratuti.
O que é Biologia Sintética?
 Este post deveria ser um dos primeiros artigos para um site que se diz especializado em biologia sintética. Mas confesso para vocês que definir o que é biologia sintética, para mim, não foi (é) uma tarefa fácil. Como podem ser vistos nos posts no blog, existem vários aspectos da synbio que permitem diferentes definições de acordo com o ponto de vista de quem está fazendo biologia sintética. Por exemplo, um engenheiro interessado em criar dispositivos computacionais sintéticos em uma bactéria, ou um biotecnólogo interessado em produzir biocombustíveis ou um biólogo querendo montar uma bactéria a partir de simples elementos químicos. Claramente, todas estas áreas estão conectadas, mas criar uma definição que consiga embarcar todas as possibilidades não é trivial. Por isso, digo que este post é orgânico, que deve mudar à medida que o meu conhecimento sobre o assunto se aprofunda.
Este post deveria ser um dos primeiros artigos para um site que se diz especializado em biologia sintética. Mas confesso para vocês que definir o que é biologia sintética, para mim, não foi (é) uma tarefa fácil. Como podem ser vistos nos posts no blog, existem vários aspectos da synbio que permitem diferentes definições de acordo com o ponto de vista de quem está fazendo biologia sintética. Por exemplo, um engenheiro interessado em criar dispositivos computacionais sintéticos em uma bactéria, ou um biotecnólogo interessado em produzir biocombustíveis ou um biólogo querendo montar uma bactéria a partir de simples elementos químicos. Claramente, todas estas áreas estão conectadas, mas criar uma definição que consiga embarcar todas as possibilidades não é trivial. Por isso, digo que este post é orgânico, que deve mudar à medida que o meu conhecimento sobre o assunto se aprofunda.
Uma característica que une todos os biológos sintéticos é a vontade de tornar o processo de engenharia de sistemas biológicos mais fácil e confiável. Dentro desse contexto, existem quatro diferentes níveis de atuação da biologia sintética:
(i) partes biológicas: sendo o DNA a linguagem de programação, as partes biológicas são uma sequência de dados (AGCTA…) que possuem funções determinadas. Por exemplo, uma sequência de DNA que faz a célula mudar a cor de verde para amarelo. Estas partes são descritas, catalogadas e respeitam determinado padrão físico de montagem (leia mais sobre os biobricks). Espera-se que com o tempo se possam descrever as características de inúmeras partes biológicas para serem utilizadas para a construção de dispositivos, sistemas,…. Essa é uma das funções da Secretária de Partes Biológicas Padrão do MIT.
(ii) dispositivos sintéticos: são compostos por partes biológicas capazes de processar sinais. Processam inputs em outputs. Para a construção de dispositivos robustos e eficientes são necessárias partes que funcionem de uma maneira previsível. Veja mais sobre dispositivos sintéticos.
(iii) sistemas sintéticos: são um conjunto dispositivos capazes de captar sinais, processar informações e realizar funções determinadas, como por exemplo, uma célula capaz de captar algum sinal do ambiente e decidir se irá realizar uma determinada função como combater uma célula tumoral, produzir determinado metabólito etc.
(iv) por útimo, existe a arquitetura sintética de populações em que, por ex, cada microrganismo possui um dispositivo diferente, sendo necessário que estes dispositivos trabalhem em conjunto para realizar determinada função. Trabalhar em conjunto, como uma população que precisa trabalhar com sincronismo, é o caso dos osciladores. Para isso, é necessário dominar mecanismos robustos de comunicação célula-célula.
A biologia sintética pode ser aplicada em praticamente todas áreas da biologia molecular, biotecnologia e engenharia genética. Porém existem algumas áreas que se destacam como sendo próprias da biologia sintética:
1. Construção de uma célula mínima: identificação das partes básicas para construção de uma célula.
2. Reconstrução de células: tendo como objetivo central a construção de formas de vida artificiais a partir de elementos químicos.
3. Construção de novos códigos genéticos.
5. Construção de células capazes de realizar funções diferentes daquelas encontradas na natureza, como a obtenção de novas rotas bioquímicas de produção de novos compostos. .
Essa última, talvez seja a que mais tenha impacto nas nossas vidas cotidianas a curto-prazo, através do desenvolvimento de remédios mais baratos e com a produção de combustíveis e químicos utilizando recursos renováveis.
Problemas com PCR!
Após desenhar o seu primer, montar a sua reação e programar o termociclador, você pode enfrentar problemas para otimizar as sua condições de PCR. Algumas simples ações podem resultar em um produto de PCR específico. Estas são as questões mais comuns aqui no laboratório:
1. Estou tendo (muitos) fragmentos inespecíficos maiores do que o esperado. O que fazer?
– Diminuir o tempo de anelamento da reação;
– Aumentar a temperatura de anelamento (aumento 2 graus a cada teste);
– Diminuir o tempo de extensão;
– Colocar menos primers;
– Checar e colocar menos DNA;
2. Estou obtendo fragmentos inespecíficos menores do que o esperado. O que fazer?
– Aumentar a temperatura de anelamento;
– Aumentar o tempo de anelamento;
– Aumentar a temperatura de extensão;
– Aumentar a temperatura de extensão para 74-78 oC;
– Colocar menos primers e/ou menos DNA.
3. A reação estava funcionando antes, mas agora eu não obtenho nenhum produto.
– Tenha certeza que todos os componentes estão na reação (tampão, DNA, primers…);
– Teste um novo Master Mix ou uma nova solução de dNTP (são sensíveis a descongelamentos consecutivos);
– Utilize um estoque novo de primers;
– Diminua sua temperatura de anelamento 6-10 oC, se não obtiver nenhum fragmento verifique todas os componentes.
Nos próximos posts vou comentar sobre técnicas de PCR: como RT-PCR, deleção de genes…
Como desenhar um primer? E como montar uma reação de PCR?
 No post passado eu comentei sobre a técnica de PCR e de como programar um PCR. Neste post eu vou um pouco mais adiante e vou falar sobre como desenhar um iniciador (primer) e como montar uma reação de PCR.
No post passado eu comentei sobre a técnica de PCR e de como programar um PCR. Neste post eu vou um pouco mais adiante e vou falar sobre como desenhar um iniciador (primer) e como montar uma reação de PCR.
O desenho do primer tem que ser feito com muito cuidado para garantir o futuro do projeto, erros no desenho podem ser percebidos apenas muito tempo depois, após muito esforço e dinheiro já ter sido gasto no projeto. Por isso, realize essa tarefa com muito cuidado, tendo em vista todo o projeto.
A primeira coisa é pegar a sequência de interesse no GenBank, como por exemplo para o gene codificador da enzima xilose isomerase de Escherichia coli. Conheça o gene, busque artigos sobre ele, veja os genes que estão envolta, como ele é regulado,… Depois salve a sequência do gene e mais ou menos 200 bp envolta dele em um documento word. Encontre o start códon, o stop códon, promotor,… e decida a sua estratégia de clonagem.
Para desenhar primers para amplificar o gene inteiro você precisará de regiões envolta do gene. Além disso existem uma série de regras a que você precisa ficar atento para desenhar o primer como: composição de G-C, tamanho do primer, temperatura de anelamento, formação de dímeros e estruturas secundárias e muito mais. Para este trabalho, eu recomendo utilizar um software, o FastPCR. Um software livre (gratuito) e amigável.
É preciso apenas colocar a sua sequência de interesse e pedir para que ele desenhe os primers, depois você precisa checar se aquela sequência amplifica a sua região de interesse. Para a encontrar o primer reverso na sua sequência tem uma ferramenta para obter o reverso complementar da sequência, já que as sequencias de primers são dadas no sentido 5´ – 3´.
Finalmente, você pode fazer um PCR in silico, para isso, volte para o Genbank e copie toda a sequência do genoma da bactéria desejada (isso pode demorar alguns minutos) e passe para o FastPCR para fazer o PCR in silico com os primers desenhados. Com isso, você pode verificar possíveis regiões amplificadas pelo seu par de primers.
Depois de mandar sintetizar os primers, você pode fazer um programa como mostrado no post anterior. Para a reação PCR você deve seguir o protocolo da sua Taq polimerase comprada. Vale a pena ressaltar que para amplificações de genes que serão expressos posteriormente, recomendo uma Taq polimerase de alta fidelidade.
No próximo post vou comentar sobre erros e soluções comuns que ocorrem com reações de PCR.
O que é um PCR? Como montar um programa de PCR?
Polymerase chain reaction ou reação da polimerase em cadeia (PCR) é técnica de biologia molecular utilizada para amplificar (aumentar o número) de um ou de alguns pedaços de DNA em várias ordens de magnitude, gerando milhões de cópias de uma sequência particular de DNA.
Utilizando um termociclador (aparelho que permite mudanças rápidas de temperatura), uma DNA polimerase, um par de iniciadores (primers) e dNTP´s (deoxinucleotídeos, que formarão a nova molécula de DNA) é possível sintetizar in vitro DNA a partir de uma fita molde. Veja o vídeo no youtube.
Diz a lenda que Kary Mullis teve a idéia do PCR em um daqueles momentos de ócio produtivo enquanto surfava nas ondas do Sul da Califórnia. Um ano mais tarde, 1983, a sua idéia virou realidade através do desenvolvimento da primeira máquina de PCR. Dez anos mais tarde, 1993, Mullis também foi surfar após receber a notícia que havia sido ganhador do prêmio Nobel pela sua invenção. Essa é uma ótima desculpa para não trabalhar aos finais de semana no laboratório! (rs)
A partir do programa de PCR básico abaixo é possível desenhar um programa específico para seu iniciador e fragmento a ser amplificado. No passo (step) 2, ocorre a denaturação do DNA, No passo 3, ocorre a ligação do iniciador ao seu DNA, para isso é preciso calcular a temperatura de anelamento do seu iniciador. Existe um cálculo baseado na composição dos iniciadores (Ta = 2(A + T) + 4(C + G) – 5), mas existem alguns softwares que calculam para você (porém esta temperatura não deve ser abaixo de 50oC, para evitar ligações inespecíficas). Após a ligação dos iniciadores ao DNA, irá ocorrer a síntese de DNA pela DNA Polimerase, normalmente se utiliza a Taq polimerase que possui uma temperatura ideal de 72oC. O tempo de elongamento da sua sequência depende do tamanho dela, calcula-se cerca de 1 min para cada 1000 pares de base (não menos de 30 segundos). Finalmente, este ciclo será repetido 30 vezes.
Programa básico de PCR:
Step Temperature (°C) Time (min)
1 95 2:00
2 95 0:30
3 55 0:30
4 72 1:00
5 Go to step 3, 30 times
6 4 forever
No próximo post vou comentar sobre o desenho de iniciadores (primers) e como montar uma reação de PCR!