


CRISPR: nova e revolucionária técnica para edição de genoma
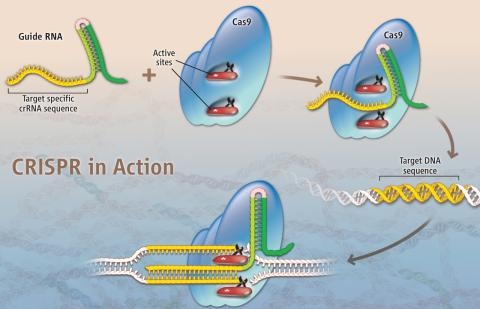
 Ter o genoma sequenciado por apenas 1000 dólares em breve será uma realidade. E não é somente ler o genoma que está se tornando cada vez mais acessível, descobertas recentes resultaram em uma nova ferramenta de edição de genoma que promete revolucionar a pesquisa médica e o tratamento de algumas doenças. Este mecanismo, chamado de CRISPR, é baseado num sistema de defesa contra vírus, uma espécie de sistema imunológico, encontrado em bactérias. E mais uma aprendemos algo interessante com estes seres unicelulares (discutimos num post anterior como bactérias podem ajudar a combater o cancer).
Ter o genoma sequenciado por apenas 1000 dólares em breve será uma realidade. E não é somente ler o genoma que está se tornando cada vez mais acessível, descobertas recentes resultaram em uma nova ferramenta de edição de genoma que promete revolucionar a pesquisa médica e o tratamento de algumas doenças. Este mecanismo, chamado de CRISPR, é baseado num sistema de defesa contra vírus, uma espécie de sistema imunológico, encontrado em bactérias. E mais uma aprendemos algo interessante com estes seres unicelulares (discutimos num post anterior como bactérias podem ajudar a combater o cancer).
Editando o genoma para curar doenças
Tornar o tratamento de desordens genéticas é sem dúvida uma das mais excitantes possibilidades desta nova técnica, principalmente disordens causadas por uma ou poucas mutações, tais como doença de Huntington. Para comprovar que isto pode ser feito, cientistas do MIT, em experimentos em camundongos, conseguiram curar em uma doença rara que ataca o fígado e é causado pela mutação de apenas um par de base de DNA. Esta doença, que também ocorre em humanos, afeta 1 em cada 100.000 pessoas e consiste na falha da quebra do aminoácido tirosina que acumula e afeta o funcionamento do fígado. Utilizando a técnica de CRISPR os cientistas conseguiram corrigir o gene para 1 em cada 250 células do figado (hepatócitos) dos camundongos. Depois de 30 dias estas células proliferaram e substituiram parte das células com o gene defeituoso chegando a um terço da população total de células, o que foi suficiente para curar a doença. Veja o artigo publicado na Nature.
Mecanismo básico das ferramentas de edição de genoma
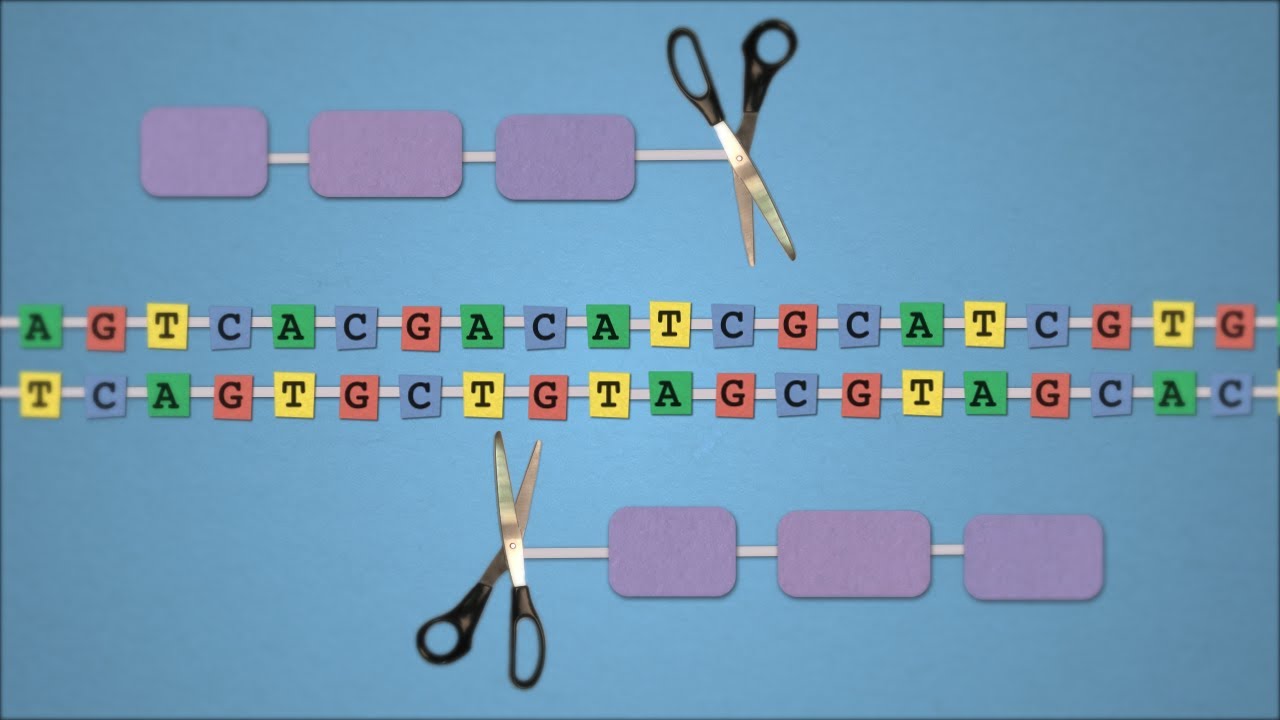
 Basicamente, o mecanismo de edição de genoma consiste em um sistema para reconhecer o sítio onde haverá a mudança combinado a um mecanismo de corte do DNA (nucleases). Uma vez reconhecido o local de corte as nucleases agem fazendo um corte nas duas fitas do DNA. Uma vez cortado, mecanismos de reparação do genoma tendem a juntar as fitas novamente e neste processo um pedaço de DNA pode ser removido ou até mesmo trocado por outro pedaço de DNA.
Basicamente, o mecanismo de edição de genoma consiste em um sistema para reconhecer o sítio onde haverá a mudança combinado a um mecanismo de corte do DNA (nucleases). Uma vez reconhecido o local de corte as nucleases agem fazendo um corte nas duas fitas do DNA. Uma vez cortado, mecanismos de reparação do genoma tendem a juntar as fitas novamente e neste processo um pedaço de DNA pode ser removido ou até mesmo trocado por outro pedaço de DNA.
As primeiras técnicas desenvolvidas, tanto Zinc finger nucleases quanto TALEN, utilizam proteínas para reconhecer o sítio de corte no genoma. Proteínas são pesadas e díficeis de projetar, diferentemente de RNA que pode ser facilmente sintetizado. E é aí que está a grande inovação da técnica de CRISPR, em utilizar pequenos pedaços de RNA para identificar o sítio de corte, o que torna a técnica simples e de baixo custo.
Recomendo os seguintes vídeos/animações para uma ilustração do mecanismo de edição de genomas. O primeiro video (em inglês) fala um pouco sobre os mecanismos gerais destas técnicas. O segundo vídeo (também em inglês) ilustra o mecanismo baseado na CRISPR.
Referência:
Bactérias na guerra contra o câncer
 Vimos num post anterior, que bactérias idênticas em seu DNA podem tomar diferentes decisões quando estão sobre stress tais como escassez de alimento. Diversas estratégias tais como ficar dormentes (esporulação), entrar em competência ou até mesmo canibalismo são utilizadas para aumentar as chances de sobrevivência da colônia. Nos últimos anos, tem aumentado o número de evidências de que células cancerígenas agem de maneira bastante semelhante. Utilizando um avançado sistema de cooperação e comunicação celular, estas células são capazes de se espalhar pelo corpo colonizando novos órgãos (metástase) ou resistir a ações clínicas tais como quimioterapia. Uma das maneiras de resistir a quimioterapia, por exemplo, é pela estratégia de tornar-se “dormente” adotada por algumas das células do tumor, processo análogo a esporulação das bactérias.
Vimos num post anterior, que bactérias idênticas em seu DNA podem tomar diferentes decisões quando estão sobre stress tais como escassez de alimento. Diversas estratégias tais como ficar dormentes (esporulação), entrar em competência ou até mesmo canibalismo são utilizadas para aumentar as chances de sobrevivência da colônia. Nos últimos anos, tem aumentado o número de evidências de que células cancerígenas agem de maneira bastante semelhante. Utilizando um avançado sistema de cooperação e comunicação celular, estas células são capazes de se espalhar pelo corpo colonizando novos órgãos (metástase) ou resistir a ações clínicas tais como quimioterapia. Uma das maneiras de resistir a quimioterapia, por exemplo, é pela estratégia de tornar-se “dormente” adotada por algumas das células do tumor, processo análogo a esporulação das bactérias.
Atualmente, desvendar o sistema de comunicação utilizado por células cancerígenas tem sido foco de inúmeras pesquisas. Como em qualquer guerra moderna, destruir o sistema de comunicação inimigo pode causar grandes danos. E na guerra contra o câncer provavelmente não será diferente. Impedir as células de se comunicarem pode evitar que elas adotem estratégias inteligentes tais como ficarem dormentes durante quimioterapia ou até mesmo matar células irmãs para obtenção de alimento. Uma vez entendido a linguagem utilizada por estas células, podemos utilizar isto ao nosso favor, interferindo nas mensagens e fazendo com que células dormentes sejam acordadas durante a quimioterapia ou até mesmo induzir as células cancerígenas a matarem umas as outras.
Finalmente, uma possível estratégia futura seria recrutar bactérias para derrotar o câncer. Elas poderiam ser utilizadas para “ensinar” as células do sistema imunológico a reconhecer e matar as células cancerígenas. É importante ressaltar que ainda compreendemos muito pouco sobre os mecanismos envolvidos no câncer e novas abordagens são necessárias para superá-lo. Entretanto, quem sabe num futuro próximo, estaremos em uma era de guerra cibernética biológica onde bactérias serão inteligentemente projetadas para derrotar o câncer.
Referências:
“Bacterial survival strategies suggest rethinking cancer cooperativity” Eshel Ben-Jacob, Donald S. Coffey, Herbert Levine. Trends in Microbiology. 2012.
“Bacterial linguistic communication and social intelligence” Eshel Ben-Jacob, Israela Becker, Yoash Shapira, Herbert Levine. Trends in Microbiology. 2004
Transposons: pedaços de DNA que mudam de endereço no genoma.
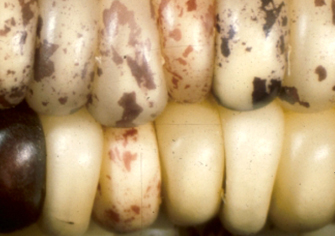
Mudança no padrão das cores do milho devido a interação entre os genes responsáveis pelo pigmento e elementos transponíveis.
Você sabia que alguns trechos de DNA têm a capacidade de mover-se no genoma, saindo de um cromossomo e se inserindo em outro? Pois é, e esta descoberta foi feita há muito tempo atrás por uma brilhante cientista, antes mesmo da descoberta da estrutura do DNA por Watson e Crick. Barbara McClintock ao observar a relação entre os padrões das cores do milho e algumas quebras cromossomais, percebeu que algumas destas quebras ocorriam com uma freqüência muito mais alta do que o esperado e, surpreendente, sempre no mesmo cromossomo. Após uma série de experimentos, a cientista percebeu que esta quebra ocorria devido a inserção de pedaços de DNA vindos de outros cromossomos que alteravam a expressão dos genes de pigmento. Confiante nos seus experimentos, Barbara então propôs que havia elementos transponíveis no genoma, ou seja, os genes não tinham um endereço fixo, mas tinham um mecanismo para movimentar-se dentro do genoma.
Se nos dias de hoje esta ideia parece bastante surpreendente, imagine como foi a recepção desta ousada ideia no começo da década de 50, quando a cientista começou a publicar seus resultados. Eles foram ignorados e ridicularizados pelos seus contemporâneos, levando a cientista a parar de publicar suas descobertas sobre o tema. Somente no início da década de 70 alguns elementos transponíveis foram identificados em bactéria validando assim a teoria de Barbara. Entretanto, o reconhecimento completo de uma das mais importantes descobertas da biologia somente aconteceu no início da década de 80, quando a cientista foi homenageada com o prêmio Nobel.
Atualmente vários estudos já relacionaram os elementos transponíveis, chamados de transposons, a uma série de doenças e como grande fonte de variabilidade genética. Hoje sabe-se que boa parte do genoma é composto destes elementos e dois mecanismos principais já foram identificados. O primeiro deles é um mecanismo de recortar-e-colar, onde alguns pedaços do DNA simplesmente saem de um lugar e movem-se para outra parte do genoma. O outro mecanismo é do tipo cortar-e-colar, gerando mais de uma cópia no genoma. Este último mecanismo é chamado de retrotransposons e consiste em quase metade do genoma humano e será tema de um próximo post.
FAQ #8 Como ver se a transformação gênica deu certo?

Já colocamos nossos plasmídeos nos bichinhos, mas ainda não acabou. Essa é a hora de saber se tudo que fizemos até aqui deu certo!
Primeiro, colocamos eles na placa de seleção com antibiótico e só aqueles que realmente incorporaram o plasmídeo vão sobreviver porque ganharam um gene amigo de resistência ao antibiótico. Agora pegamos uma parte das células sobreviventes e fazemos um teste usando a PCR (você lembra dessa técnica, né?).
Como testar com a PCR?! Roubamos os plasmídeos dessas células e tentamos multiplicar o pedaço de DNA que foi grudado nele adicionando primers, nucleotídeos e enzimas sob aquecimento e desaquecimento. Se o nosso plasmídeo modificado tiver sido incorporado pelos micro-organismos haverá a multiplicação do pedacinho e podemos vê-los em uma eletroforese. Caso contrário, nada foi copiado e nada vai aparecer na eletroforese. Então temos o sinal de que alguma coisa não deu certo por aqui e as chances de erros deste teste são bem pequenas.
Depois vamos para um teste melhor ainda, o sequenciamento. Este teste diz exatamente qual é a sequência de pares de base da molécula do plasmídeo e acabam de vez suas dúvidas se a transformação deu certo ou não! O sequenciamento começa parecendo um processo de duplicação normal de DNA. Mas além de cadeias molde, enzimas e nucleotídeos normais, há nucleotídeos especiais sintetizados (ddNTP’s) que possuem duas boas propriedades: emitem luz e interrompem o prologamento da cadeia a partir de onde foram adicionados. Então, imagine uma molécula de DNA que possui um par de base A-T em um determinado comprimento e considere que a base A pertence à cadeia molde e a base T à cadeia complementar. Se esta base T for um nucleotídeo especial temos como identificar “quem é” e “qual sua posição” na cadeia, pois ele emitirá uma cor específica para Timina e o comprimento de sua cadeia está interrompido na posição exata (nº 2 em verde). 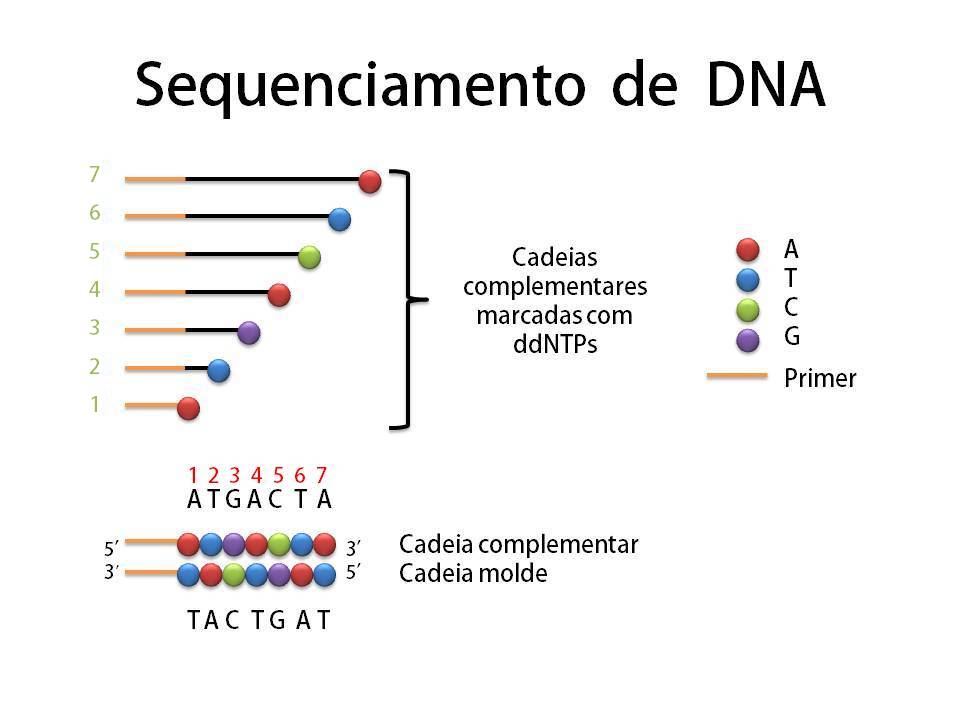 Podemos detectar a cor emitida através de um espectrógrafo e a posição pelo tamanho revelado na eletroforese em gel (o sequenciador é basicamente a união dos dois). Ou detectar o tipo de nucleotídeo presente não pela cor, mas colocando os filamentos “lavados” por cada tipo de nucleotídeo em poços diferentes do gel e interpretando a sequência a olho nu/manualmente. Depois de identificado o nucleotídeo da cadeia complementar é possível saber quem ocupa a mesma posição na cadeia molde, no caso citado é a base Adenina. Expanda esse raciocínio para todas as outras bases da molécula, com uma amostra grande teremos cadeias de todos os comprimentos possíveis, interrompidas por um dos 4 tipos de nucleotídeos especiais (A, T, C, G), e assim é possível sequenciar toda a molécula de DNA!
Podemos detectar a cor emitida através de um espectrógrafo e a posição pelo tamanho revelado na eletroforese em gel (o sequenciador é basicamente a união dos dois). Ou detectar o tipo de nucleotídeo presente não pela cor, mas colocando os filamentos “lavados” por cada tipo de nucleotídeo em poços diferentes do gel e interpretando a sequência a olho nu/manualmente. Depois de identificado o nucleotídeo da cadeia complementar é possível saber quem ocupa a mesma posição na cadeia molde, no caso citado é a base Adenina. Expanda esse raciocínio para todas as outras bases da molécula, com uma amostra grande teremos cadeias de todos os comprimentos possíveis, interrompidas por um dos 4 tipos de nucleotídeos especiais (A, T, C, G), e assim é possível sequenciar toda a molécula de DNA!
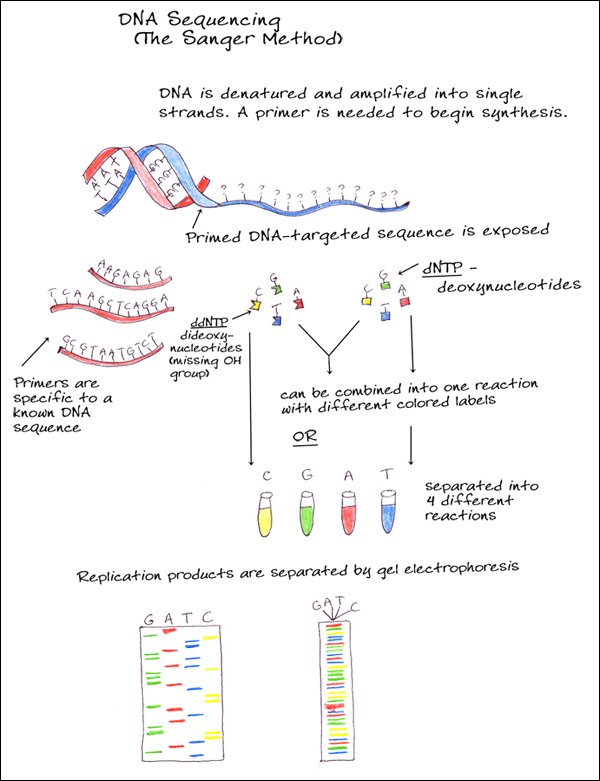
Para saber mais sobre sequenciamento genético, clique aqui.
Por Otto Heringer e Viviane Siratuti.
FAQ #7 Como enfiar o plasmídeo nas células?

Dar e receber plasmídeos não é nenhuma novidade para algumas células!
As bactérias costumam trocar informações genéticas assim e esse processo é chamado de conjugação, portanto elas já possuem toda maquinaria necessária pra isso! Desse jeito elas aumentam muito a variabilidade e as chances de sobrevivência da população.
Podemos usar esse mecanismo natural para enfiar os plasmídeos, mas na maioria das vezes damos uma ajudinha usando choques térmicos ou elétricos.
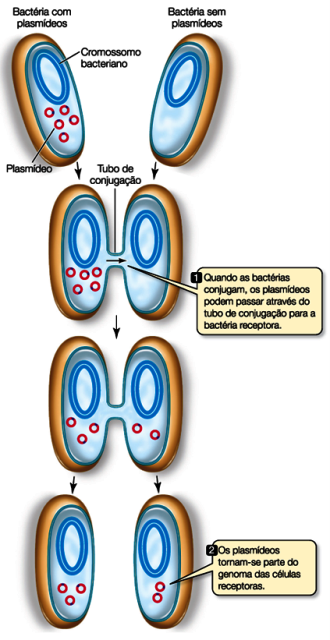
Para entender o choque térmico, assista o vídeo do JOVE aqui. E do choque elétrico, aqui.
Por Otto Heringer e Viviane Siratuti.
FAQ #6 Como ver se nossos plasmídeos incorporaram os pedacinhos de DNA?

Sabendo o tamanho do plasmídeo e dos pedacinhos estimamos um tamanho para nosso novo plasmídeo e agora é só conferir se estão como esperado!
Para isso, existe uma técnica de separação chamada eletroforese em gel, onde as moléculas são “peneiradas” por algum polímero (gel de poliacrilamida ou de agarose) quando se movimentam atraídas ou repulsadas pelos eletrodos de cargas opostas que são colocados nas extremidades do gel. Ou seja, uma molécula com carga negativa caminha para o eletrodo de carga positiva e vice-versa. E as moléculas de mesma carga correm de maneiras diferentes pelo gel de acordo com seus pesos moleculares e tamanhos, as menores e mais “leves” passam com mais facilidade pelo gel enquanto as maiores e mais “pesadas” ficam mais retidas. No final, temos as moléculas separadas ao longo do gel e podemos comparar com a ladder (uma “régua” feita de moléculas com pesos e tamanhos já conhecidos) para saber se nossos plasmídeos estão no tamanho esperado.
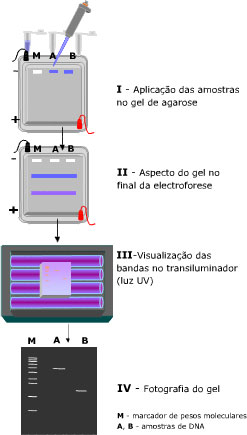
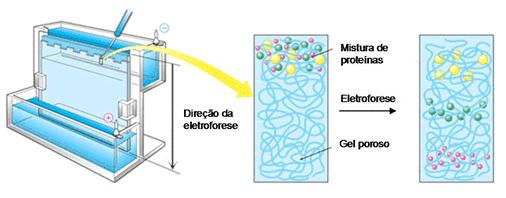
Depois de confirmar pela eletroforese que nossos plasmídeos foram modificados, precisamos “salvá-los” do gel para colocar nas células. Basicamente, cortamos o pedaço de gel que está com nossos plasmídeos e colocamos em tubinhos com uma pequena coluna de sílica dentro (imagem aí embaixo). Então as moléculas de DNA se ligam à coluna, fazemos uma lavagem para tirar o gel e depois diluímos o DNA para retirá-lo da coluna!

Para saber mais sobre eletroforese, assista o vídeo do JOVE Science aqui e purificação aqui. 🙂
Por Otto Heringer e Viviane Siratuti.
FAQ #5 Como colocar os pedaços de DNA no plasmídeo?

Para abrir os plasmídeos (lembrando que é um DNA circular) e depois grudar os pedacinhos de DNA que extraímos pela Miniprep e multiplicamos pela PCR, usamos as chamadas enzimas de restrição e de ligação. Isso tudo ainda fora das células, ok?!
As enzimas de restrição reconhecem e cortam os plasmídeos em regiões específicas que queremos, isto é, cortam em determinadas sequências de pares de base. Inclusive existem dois tipos de corte, podendo formar extremidades coesivas (cortes em comprimentos diferentes entre as duas fitas) ou cegas (corte reto na dupla fita).
Feito isso, agora usamos as enzimas de ligação para grudar as extremidades dos plasmídeos às extremidades dos pedacinhos, onde as bases se complementam.
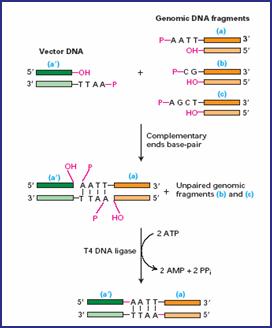
Legal saber também que o nome dessas enzimas estão sempre relacionados com os nomes dos organismos onde elas foram encontradas (veja a enzima EcoRI, por exemplo).

Para assistir o vídeo do JOVE Science sobre enzimas de restrição, clique aqui. E enzimas de ligação, aqui.
Por Otto Heringer e Viviane Siratuti.
FAQ #4 Onde colocar esses pedaços de DNA?

“No genoma, né, dêr!”. Certo, mas como? E será que o genoma é o único lugar que podemos colocar esse novo pedacinho de DNA no micro-organismo que queremos modificar? Não! Existe outro lugar também e ele se chama plasmídeo (quem já jogou BioShock vai soltar umas sinapses a mais agora).

O plasmídeo é um DNA circular presente em várias espécies de seres vivos e é responsável por conter informações valiosas envolvendo a sobrevivência do organismo a fatores externos (como por exemplo sua resistência a um antibiótico), além de ser o principal ator da transferência horizontal de informação genética, ou seja, a passagem de um DNA funcional de um ser vivo para outro sem haver hereditariedade (é aí que o jogo BioShock extrapola isso para seres humanos). Adivinha de onde veio a ideia de usar o plasmídeo como transmissor – “vetor” – de informação genética para modificar as células? Veio exatamente desse mecanismo natural de realizar transferência horizontal de genes que vários micro-organismos possuem, então aproveitamos para fazer a transferência das informações genéticas que nós queremos!
E se vamos usar plasmídeos é preciso extraí-los também! Os plasmídeos são moléculas de DNA assim como os pedacinhos obtidos a partir de um genoma, e multiplicados pela PCR, que vamos introduzir no microorganismo, mas aqui há uma etapa importante durante a extração de DNA onde é feita a separação do conteúdo plasmidial do genômico.
O vídeozinho abaixo tem uma animação no ínicio e depois mostra o procedimento do isolamento do plasmídeo em lab!
[youtube_sc url=”http://www.youtube.com/watch?v=8xEDEJ0DHFA”]
Por Otto Heringer e Viviane Siratuti.
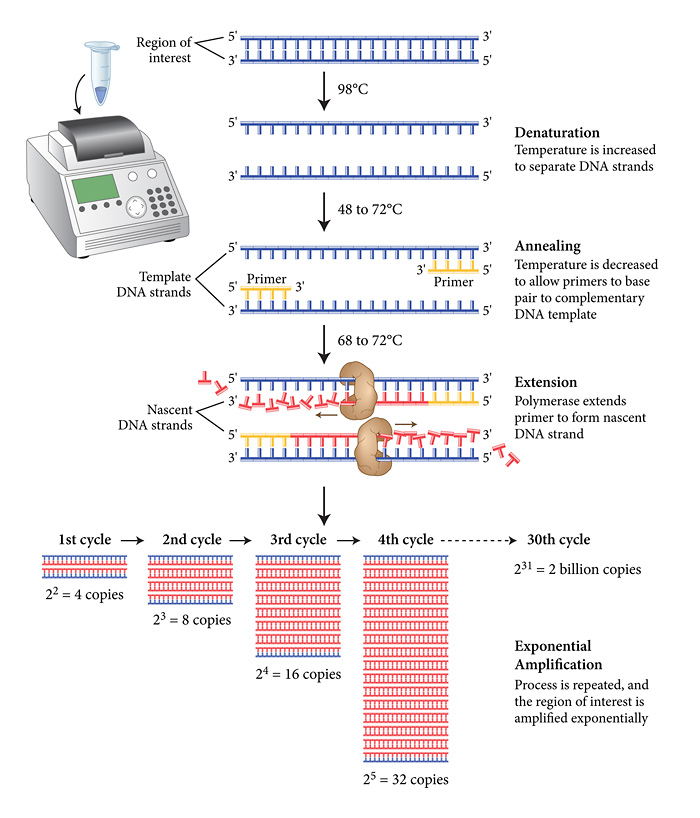
FAQ #3 Como copiar o código em laboratório?

Ahá! Essa é a pergunta que mudou a biotecnologia. Até conseguirmos fazer isso, nós (i.e. humanidade) passamos por um caminho bem interessante envolvendo prêmios nobel e momentos epifânicos. O videozinho explica muito melhor do que esse texto corrido como funciona a metodologia pra se fazer isso, a Reação em Cadeia da Polimerase (Polimerase Chain Reaction), o tão amado e odiado (quando simplesmente não funciona) PCR!
[youtube_sc url=”http://www.youtube.com/watch?v=vmlLj1aLZ7s”]
Encurtando a história: usando uma enzima que trabalha “sentando” no molde de uma fita única de DNA, aquecimento e desaquecimento (para “ligar” e “desligar” essa enzima) e pedacinhos de nucleotídeo, conseguimos fazer milhões de cópias de apenas uma única molécula de DNA – é por isso que o pessoal do CSI (o seriado) consegue uma grande informação genética usando apenas resíduos quase desprezíveis de material biológico.
Por Otto Heringer e Viviane Siratuti.
FAQ #2 Como extrair um pedaço de DNA desejado?

Queremos pegar um pedacinho codificante de DNA (responsável por alguma característica no organismo) de um outro pedaço maior de DNA. OK, mas onde fica esse outro pedaço? Bem, existem pedaços disso praticamente a todo o seu redor. Onde há vida, há informação genética que pode ser “pega”. Extraímos ele basicamente fazemos lavagens usando solventes de diferentes “afinidades de dissolução” (a grosso modo) com as biomoléculas da célula até restar o DNA.
Se você quer saber, existem até kits comerciais para se fazer isso (como por exemplo a imagem abaixo), são os famigerados “kits de miniprep”. “Mini” porque em geral são bastante usados kits que trabalham com pequenos volumes (microlitros), mas também existem os kits de “midiprep” e “maxiprep”, para volumes maiores.

Assista o vídeo caso você queira saber mais do processo (aviso: a realidade às vezes não é algo tão excitante como você imaginava).
[youtube_sc url=”http://www.youtube.com/watch?v=P12DPa-g8Ro”]
Aliás, se você ficou com vontade de extrair DNA em casa, é bem simples, clique aqui.
Por Otto Heringer e Viviane Siratuti.