


FAQs da Bioengenharia – Introdução

Preocupados com os novos amantes da biologia sintética, estamos procurando materiais de introdução no assunto e encontramos algumas coisas interessantes! 🙂
Por isso vamos colocar uma sequência de posts explicativos, partindo de uma visão geral e seguindo por perguntas mais específicas. É um pequeno tutorial-FAQ sobre alguns fundamentos que quando entendidos ajudam bastante a entender algumas de nossas discussões e outros posts deste blog. Serve para ajudar principalmente quem não é da área de biológicas ou quem ainda não se aprofundou muito neste assunto! Se continuarem com dúvidas, perguntem tá?!
Lembrem-se de ativar as legendas dos vídeos, caso necessitem! Todos possuem legendas originais em inglês e alguns em português. Você pode colocá-las traduzidas quando só houver em inglês (não muito recomendado, rs). Nos vídeos do JOVE existe a opção de colocar os textos em português. Se você não sabia que o YouTube fazia isso por você, dá uma olhada aqui (dica: barrinha inferior do vídeo).
Antes de mais nada, você sabe o que são e quais as relações entre nossas principais moléculas orgânicas (DNA, RNA e proteínas), certo? Se sua resposta é não, assista um desses vídeos!
[youtube_sc url=”http://www.youtube.com/watch?v=h3b9ArupXZg#t=0″]
[youtube_sc url=”http://www.youtube.com/watch?v=tMr9XH64rtM”]
Indo além da Estrutura
OK, agora vamos além desses conceitos básicos dos vídeos. Mais que entender a estrutura e a dinâmica dessas biomóleculas, como os cientistas a alteram? Em linhas gerais, como se faz para modificar geneticamente um organismo?
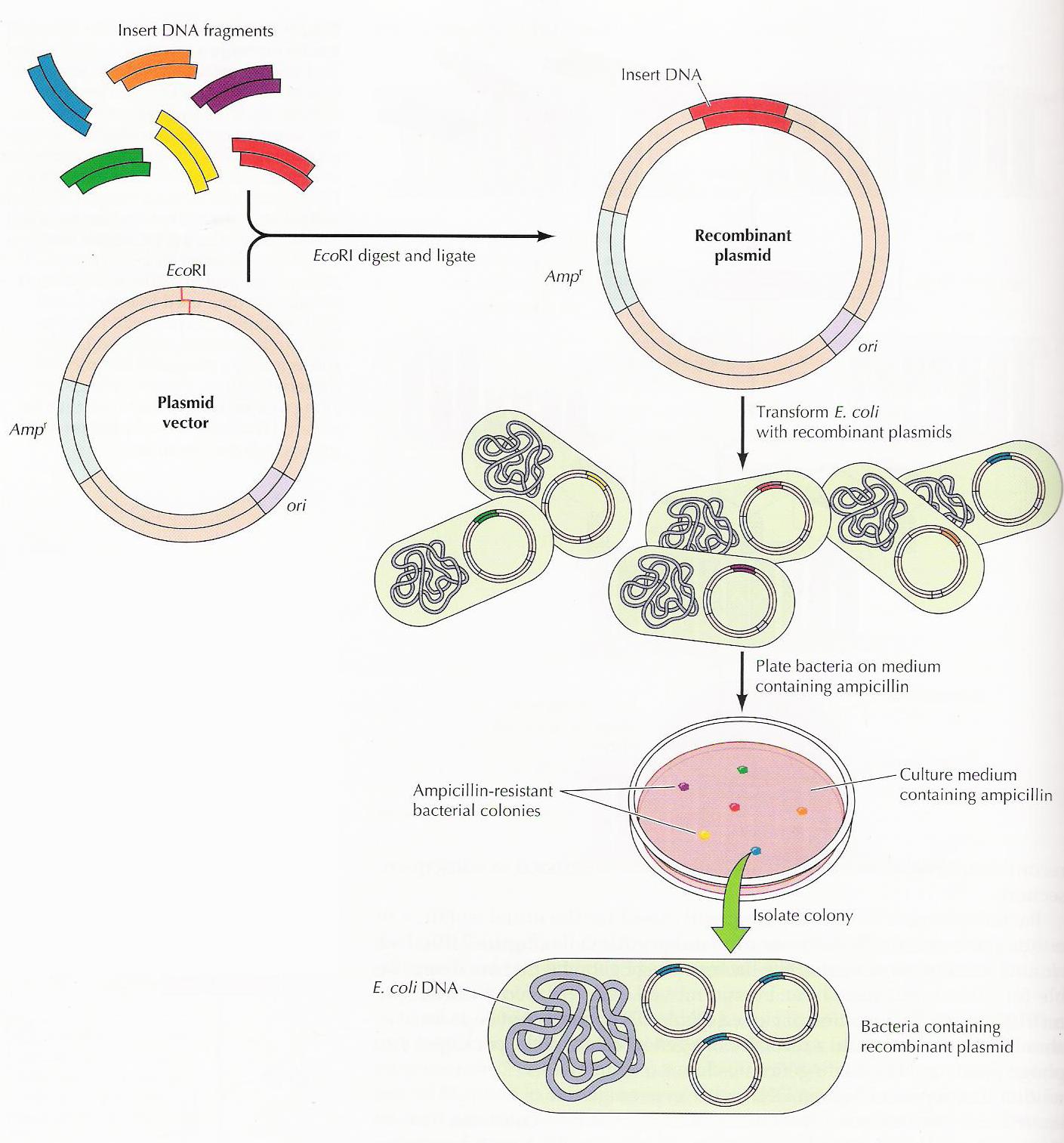
Vamos lá! Primeiro pegamos um pedacinho codificante de DNA (gene) de outro organismo que tenha certa característica que desejamos. Para isso, extraímos o DNA e depois multiplicamos só esse pedaço que nos interessa através de uma técnica chamada PCR (afinal, a eficiência das transformações é pequena e quanto mais material melhor). Precisamos também extrair e abrir os plasmídeos (DNA circular) do organismo escolhido para receber este gene e assim receber as novas características (já que o plasmídeo é nosso principal veículo de introdução dos genes nas células). Para abrir estes plasmídeos usamos enzimas de restrição que cortam as moléculas de DNA em lugares específicos e chamamos isso de digestão. Depois que tivermos vários plasmídeos e genes, podemos grudá-los com a ajuda de enzimas de ligação. E então, para saber se nosso novo plasmídeo deu certo usamos uma técnica de separação por carga e tamanho de molécula, a eletroforese em gel. Nela, as moléculas se acumulam em certas posições que nos dizem seus tamanhos (mergulhadas em um polímero) e assim podemos identificar aquelas que correspondem ao que estimamos. Além disso, fazemos testes com a técnica PCR e o sequenciamento genético. Se os testes disserem que tudo que fizemos deu certo, recuperamos o material e colocamos nossos novos plasmídeos nos organismos, geralmente utilizando choque térmico ou elétrico. Esse passo, introdução de plasmídeos, é a chamada transformação. Mas para selecionarmos somente aqueles organismos que realmente foram transformados usamos antibióticos que matam as células que não ganharam os nossos novos plasmídeos, já que não possuem os genes de resistência que foram colocados neles. E no final, cada célula que sobreviveu se multiplicará, formando colônias de organismos geneticamente modificados. Agora dá uma olhada no esquema de novo e vê se entendeu até aqui!
Por Otto Heringer e Viviane Siratuti.
Um jogo para acabar com preconceitos
Qual é a melhor maneira de passar uma informação pra uma pessoa!?
Como os comerciais, filmes e canais de televisão estão aí pra comprovar, o entretenimento passa muito mais pra você do que mera diversão. É com essa ideia que ficamos pensando em como fazer as pessoas entenderem os conceitos e finalidades da abordagem da Biologia Sintética. Como não perdemos tempo para arrumar uma desculpa para nos divertir, criamos durante esse ano um jogo de cartas – inspirado em elementos de Munchkin, Bohnanza, Magic e War – para, além de ensinar de uma maneira divertida sobre conceitos de microbiologia e biologia molecular, informar melhor as pessoas e acabar com certos preconceitos envolvendo microrganismos bioengenheirados.
E olha que legal: além de levarmos essa ideia como nossa Human Practices na competição internacional de máquinas geneticamente modificadas desse ano (e sermos bastante elogiados por esse trabalho), emplacamos primeiro lugar com o projeto na Olimpíada USP do Conhecimento!

É, senhora Sociedade, eu te disse que nossa brincadeira é uma brincadeira séria! Tão séria que esse projeto não para aqui.

O jogo estará disponível para download (se você quiser imprimir aí na sua casa) ou para compra através do maravilhoso site “The Game Crafter“, que é de uma empresa que imprime e vende jogos independentes, como o nosso. Desse jeito nosso jogo vai poder sempre fazer o que ele se propõe a fazer: ser jogado!
O jogo
O jogo funciona assim: cada jogador (até 4) escolhe uma carta de personagem personagem, como por exemplo o professor Fujita:

OBS: procure o “easter egg”.
Como dá pra ver, cada pesquisador tem uma personalidade específica e um chassi com que desenvolve seus projetos. No caso o senhor Fujita é um pesquisador que não colabora muito mas bastante competente, trabalhando com a largamente usada Escherichia coli.
O grande objetivo do jogo é construir primeiro que o seu colega um circuito gênico – afinal estamos falando de academia, minha gente! Para construir o circuito o jogador deve “criar”, acumular e trocar BioBricks, até que tenha a combinação de Biobricks necessários para completar o circuito, como por exemplo esse:

OBS: nem todos os objetivos realmente podem ser feitos em alguns chassis.
Os Biobricks podem ser baixados com “pontos de metabolismo”, que é a representação dos recursos metabólicos e energéticos que o microrganismo tem para passar com sucesso pelo processo de transformação gênica de cada parte, a ser inserida sequencialmente na célula (no exemplo anterior há 8 BioBricks).
A dinâmica das cartas se dá quando elas ainda estão na sua mão e não foram “baixadas” no organismo. Há também (no melhor estilo Munchkin – quem já jogou sabe do que estou falando!) cartas dinâmicas usadas por um jogador em si mesmo ou em outros jogadores, como essa abaixo:

E, por último, o último elemento do jogo é a tão temida aleatoriedade! Aquelas variáveis sem controle que sempre fazem seu experimento não sair como você queria. Um jogador no final da rodada joga um dado: dependendo do número tirado uma “carta aleatória” surge, ajudando ou prejudicando o ganho de pontos de metabolismo (que ocorre por rodada) dos chassis de cada pesquisador.

Fizemos um overview do projeto num vídeo do youtube, dê uma olhada:
[youtube_sc url=” http://youtu.be/6Odd5-OKyHA”]
Quando o nosso novo site ficar pronto vamos ter um endereço especial com o jogo, por enquanto fica aqui nossa promessa de acesso aberto a esse conteúdo. 🙂
Acontece nos filmes, acontece na vida, acontece no Clube de Biologia Sintética
Este é mais um projeto que surgiu das reuniões do Clube de Biologia Sintética, feito por pessoas das mais diversas áreas e que se conheceram no clube. Esse é o objetivo principal do grupo: Reunir e ensinar pessoas de maneira divertida , integrar áreas, criar projetos científicos inovadores e criativos e, por fim, gerar impactos positivos na sociedade.
Você que compartilha dos nossos ideais, acompanhe nossas reuniões pessoalmente ou pelo ao vivo pelo streaming no nosso canal do youtube, ou ainda entre em contato pelo nosso email, canal do facebook e twitter!
Estratégias para tempos adversos.

Em tempos adversos é comum haver muitas mudanças e isto não acontece somente com os humanos que saem as ruas, fazem guerra ou revolução. Na natureza observamos inúmeras estratégias dos seres vivos para superar tempos difíceis, algumas bastante chocantes do ponto de vista humano, tais como infanticídio ou canibalismo. E no mundo microscópio não é diferente.
Se você pensa que as bactérias morrem pacíficas quando as condições não são favoráveis você está bastante enganado. Até mesmo para estes minúsculos seres unicelulares a vida pode ser complexa e cheias de decisões difíceis e estressantes a serem tomadas. Existem diversas estratégias para lidar com tempos difíceis, tais como escassez de alimentos e danos ao DNA. Uma das estratégias mais radicais e mais utilizadas quando a coisa está realmente feia é formar esporos, uma resposta celular bastante complexa que envolve a ativação de mais de 500 genes ao longo de aproximadamente 10 horas. Este processo termina com a morte da célula mãe e a formação de uma célula filha dormente com a capacidade de resistir a situações extremas como calor, radiação e presença de substancias químicas. Por outro lado, durante este tempo o esporo não pode tirar vantagem imediata de situações favoráveis para se reproduzir.
Esporulação é uma tomada de decisão bastante complexa e que não se inicia simplesmente com a escassez de alimentos, mas é resultado de uma série de passos que podem ser descritas como decisões celulares sobre como lidar com o stress presente. Envolve, por exemplo, uma comunicação entre as bactérias da mesma colonia por um mecanismo chamado de quorum sensing. Além disto, antes da esporulação, comumente as bactérias tentam outras táticas como ativar um flagelo para buscar alimentos, secretar antibióticos e outras substancias na tentativa de destruir outros micróbios competidores. As células também checam uma série de condições internas antes de decidir esporular tais como a integridade do DNA cromossomal.
Mesmo quando a maior parte da colonia decide por esporular, o material gerado pela lise da membranas das células que esporularam são aproveitados por outras células da colonia que, no caminho para esporular, entram no estado chamado de competência, que consiste em abrir poros na membrana para facilitar a entrada de DNA exógeno, que pode ser utilizado para reparo do DNA e eventualmente como fonte de informação genética que as ajudará a resistir ao momento. Há ainda um outro caminho mais radical tomado por algumas células da colônia que consiste em secretar alguns fatores antibacterianos que faz com que células irmãs fiquem incapazes de esporular, causando inclusive a lise de sua membrana, em uma espécie de canibalismo. Interessante notar que, sobre a perspectiva de teoria de jogos, a estratégia de canibalismo seria predominante sobre a estratégia de entrar em competência, o que não é observado. Isto indica que possivelmente as células em competência são imunes ao canibalismo, mas não sabemos ainda se é este o caso.
Modelos teóricos/moleculares para este tipo de tomada de decisões normalmente consistem na integração de módulos gênicos formados principalmente por circuitos regulados por diversos fatores de transcrição e micro RNAs. Estes tipos de sistemas gênicos também desempenham um papel importante no desenvolvimento embrionário e de células cancerígenas, motivo pelo qual tem se dado muita atenção a este tipo de estudo.
Referencias:
Brincando de Comparar Códons
Sou daquelas pessoas que simplesmente não conseguem dormir direito com um mistério. Essa é uma obsessão que provavelmente muitos cientisas (e “wannabe scientists”, como eu) têm. Às vezes ficamos obcecados com uma coisa muito importante, às vezes com uma coisa banal e muitas vezes com algo que você nunca parou para pensar direito. O mais emocionante é que qualquer resposta de uma dúvida tem aquela probabilidade mágica de revelar algo impressionante ou bem útil. Hoje (dia dessa postagem), fiquei obcecado por tentar entender “na prática” qual é a grande ideia da otimização de códons e o quanto os organismos podem ter preferências de códons diferentes. Aqui está o registro da investigação do pequeno mistério de hoje!
Códons, Otimizações e Preferências
Antes de discorrer sobre o que andei brincando. Uma pequena contextualização ao intrépido viajante sobre o que são códons, porque eles precisam ser otimizados e o que diabos é essa “preferência de códons”.
Códons são os trios de combinações de letrinhas A,T,C e G do DNA (os nucleotídeos) que, depois de transcritos a RNA (em que a grande diferença é que os “T’s” são substituídos por “U’s”), são literalmente traduzidos em aminoácidos; ou seja: três nucleotídeos codificam um aminoácido. A grande coisa dos códons é que eles são redundantes: existe mais de uma maneira de um aminoácido específico ser traduzido à partir dos trios de nucleotídeos. Os cientistas fizeram uma tabela espertinha que “decodifica” nucleotídeos em aminoácidos:

Comece lendo do centro até às bordas do círculo combinando as letras que você for olhando pelo caminho. Por exemplo: U+A+C = Tyr, abreviação de Tirosina.
Mas aí você se pergunta: “Querida Natureza, qual é o propósito disso!?”. A redundância da leitura de aminoácidos tem uma implicação muito importante na conservação do código genético; ela é a última barreira espertinha da contra mutações no DNA. Imagine que o “C” do códon UAC que traduz uma Tirosina fosse mutado e virasse um “U” (dando UAU): graças à redundância de tradução, o aminoácido Tirosina ainda continua sendo traduzido! Pra se ter uma ideia de como isso é importante, uma única substituição de aminoácidos (o que pode acontecer com uma única mutação de nucleotídeos) já pode gerar doenças (pesquise sobre Anemia Falciforme).
Enfim, concluindo: existem muitos códons que podem ser traduzidos em diferentes tipos de aminoácidos. Como existem muitas opções, diferentes organismos costumam a ter preferências por diferentes códons para traduzir aminoácidos específicos – por exemplo: nós Humanos adoramos traduzir Arginina como AGA e AGG, já uma das bactérias do nosso cocô, a E.coli, acha muito mais interessante traduzir Arginina como CGU e CGC. Vai entender esses procariotos viu!
Mas porque isso acontece? Porque evolutivamente cada espécie foi selecionada em um ambiente particular, o que implica em diferentes necessidades de estabilidade do DNA em diferentes contextos, e portanto diferentes porcentagens de C e G, e A e T no genoma. Essas porcentagens direcionam quais códons os organismos preferem.
Por causa de tudo isso, quando algum cientista vai fazer o design de um pedaço de DNA, é preciso colocar a sequência no contexto do organismo a ser utilizado, deixando os códons “otimizados” para cada ser vivo – caso contrário, os genes inseridos no organismo serão pouco ou nada expressos.
Investigando leveduras
Mais profundamente, resolvi brincar dessas coisas querendo responder uma pergunta: “O quão compatível os códigos genéticos de duas espécies de leveduras podem ser?”. No caso, Pichia pastoris e Saccharomyces cerevisiae.
Primeiramente eu entrei no “Codon Usage Database“. Procurando por Pichia e Saccharomyces, o site dá uma tabela com a frequência de se encontrar determinado códon a cada mil pares de base. Eu peguei os resultados e coloquei num site chamado “Text Diff” – ele compara dois textos e mostra as diferenças e igualdades entre os dois. Com a comparação, dei print screen e destaquei as frequências mais discrepantes entre as duas espécies de levedura, obtendo o seguinte diagrama:

Texto em vermelho: Pichia. Texto em Verde: Saccharomyces. Laranja – diferença de 4 a 5; Rosa – diferença de 6 a 9; Amarelo – diferença acima de 10; Códons circulados – frequências iguais.
Fui atrás de cada códon, procurando o que codifica. Cheguei na seguinte tabela:

Eu chamei de “eficiência de códons” o quão os códons de Pichia funcionam em Saccharomyces, tomando como “códons incompatíveis” aqueles com diferença de no mínimo 4 entre as frequências de códon em cada espécie (a marcação em amarelo na imagem de comparação das frequências) – também estou tomando como hipótese que há uma relação direta entre frequência de códon e a preferência do mesmo por determinada espécie. Cheguei nesses valores através da porcentagem do número de códons “compatíveis” (totais – incompatíveis). De 20 aminoácidos possíveis, apenas 7 seriam seus códons prontamente compatíveis.
Ambas as espécies são leveduras, e por isso eu esperava uma maior compatibilidade natural. O problema é que eu não tenho um controle para saber se a usagem de códons de cada levedura é realmente discrepante. Por isso, fiz a mesma comparação entre Pichia e E.coli. Como esses organismos são bem mais diferentes (um é eucarioto e outro procarioto), esperei uma diferença bem maior. (veja imagem abaixo)

Texto em vermelho: Pichia. Texto em Verde: E.coli. Laranja – diferença de 4 a 5; Rosa – diferença de 6 a 9; Amarelo – diferença acima de 10; Códons circulados – frequências iguais.
Legenda: Laranja – diferença de 4 a 5; Rosa – diferença de 6 a 9; Amarelo – diferença acima de 10; Códons circulados – frequências iguais.
Como esperado, dá pra ver claramente o quanto E.coli e Pichia são diferentes em comparação com Pichia e Sccharomyces. Nesse panorama, eu diria em Pichia e Saccharomyces são bem parecidas. Quanto mais comparações forem feitas mais certeza se terá do quão um organismo se parece com outro.
Otimização de Códons
Apesar de eu não ter certeza da relação direta entre frequência e preferência de códon, consegui observar coisas muito interessantes: a única inviabilidade de tradução correta entre Pichia e Saccharomyces de aminoácido é o Glutamato, em que as frequências de todas as possibilidades de códons não entram na minha classificação de “códons compatíveis” (diferença de frequência menor que 3). O resto dos códons podem ser compatibilizados entre espécies usando-se versões alternativas de códon para um mesmo aminoácio! 🙂
Quando se otimizam códons para deixar um plasmídeo compatível em diferentes plataformas, faz-se exatamente isso. O problema é que mesmo assim a expressão ainda não é ótima, então em geral prefere-se “sacrificar” a compatibilidade do plasmídeo em diferentes espécies para se ter um plasmídeo com os melhores códons em cada bichinho.
Existem vários programas que fazer essa otimização de códons rapidamente, mas em geral as empresas que sintetizam DNA já incluem isso (de graça ou não) no planejamento do plasmídeo a ser sintetizado.
Conclusão
Por fim, a conclusão que tirei disso tudo é: eu ACHO que um gene de Pichia funcionaria suficientemente bem em Saccharomyes e vice-versa. No caso de não conseguirmos sintetizar os genes que precisamos já códon-atimizados, talvez valha a pena fazer uma mistureba de DNA interespécies – mas só para as leveduras!
Aventuras em Biologia Sintética

Drew Endy
Eu considero os quadrinhos como uma das formas mais interessantes de se narrar uma história. São também uma ótima forma de divulgar ciência de uma maneira didática e divertida.
Drew Endy um dos pais da biologia sintética (e do Registry of Parts), juntamente com Isadora Deese (ambos do MIT) elaboraram o quadrinho Adventures in Synthetic Biology. E tem mais: o quadrinho foi publicado no website da prestigiada revista Nature.
Confira Adventures in Synthetic Biology e aprenda de maneira didática o que são biobrick, PoPs, entre outros conceitos básicos.
Links possivelmente interessantes:
- Bastidores da produção do quadrinho
- The cartoon guide to genetics
- TED talk do Jorge Cham, criador do sensacional PhD Comics
Espetáculo da ciência
“We’ve arranged a global civilization in which most crucial elements profoundly depend on science and technology. We have also arranged things so that almost no one understands science and technology. This is a prescription for disaster. We might get away with it for a while, but sooner or later this combustible mixture of ignorance and power is going to blow up in our faces.”
(Carl Sagan, The Demon-Haunted World: Science as a Candle in the Dark )
[youtube_sc url=”http://youtu.be/cP2VhadII84″ title=”A%20ciência%20como%20uma%20enigmatica%20caixa%20preta%20que%20ao%20ser%20manipulada%20por%20um%20misterioso%20personagem,%20o%20cientista,%20produz%20inovações%20tecnológicas.”]
Caixa preta é um conceito que designa um sistema cujos detalhes de funcionamento são desconhecidos ou mesmo ignorados. Computadores, micro-ondas, aparelho celulares são alguns dos vários exemplos de objetos que são amplamente usados pela sociedade mas que, em geral, não há uma preocupação maior por parte dos usuários com relação ao seu funcionamento.
Grande parte da população brasileira não tem acesso à educação científica, fazendo com que os avanços tecnológicos que a ciência promove apareçam aos olhos do cidadão sob uma forma não muito diferente de um espetáculo mágico. Tem-se uma sociedade industrializada derivada dos avanços na ciência e tecnologia (sem mais discussões apontarei estas duas como indissociáveis) sentada na platéia de olho apenas para o output de uma caixa preta cuja única razão de ser é a geração de mais aplicações tecnológicas. Já o enigmático e estereotipado personagem que manipula a caixa, o cientista, faz parte de uma pequena porcentagem que detém os saberes do que está por trás do funcionamento da mesma.
Apesar de atualmente haver uma maior mobilização relacionada à divulgação científica no Brasil (como o scienceblogs), maior cobertura em revistas e jornais televisivos e principalmente na internet, o quadro ainda é frágil. A mídia, não raramente, apresenta a ciência como um empreendimento espetacular, realizado por pessoas super-dotadas, gênios que nunca cometem equívocos e que fornecem informações cem por cento confiáveis. Consequência dessa mídia exagerada é uma visão distorcida de como funciona a ciência, exemplo recente (e trágico em todos sentidos) foi o ocorrido com cientistas italianos que foram condenados por não preverem um terremoto. É dado uma grande ênfase as aplicações da ciência mas o processo de sua produção, seu contexto, suas limitações e incertezas são ignorados.
Essa falta de compreensão, reflexão e educação cientifica no entanto não impede que tenhamos uma sociedade aberta à ciência (sim!!), e como vimos bem confiante no saber tecnocientífico que superficialmente lhe é apresentada. Não é incomum, por exemplo, comerciais de televisão usarem dessa confiança no conhecimento cientifico afim de ganhar os clientes, do tipo “está comprovada cientificamente a eficácia do produto…”. Ficamos diante de uma sociedade impaciente, exigente e que aguarda cada vez mais da ciência mas que muitas vezes não consegue filtrar as informações sensacionalistas e contraditórias que lhe são apresentadas (Parece milagre! Novo remédio faz emagrecer 7 a 12 quilos em cinco meses. E sem grandes efeitos colaterais!!).
A sociedade comanda direta e indiretamente o que é produzido e proposto cientificamente. A ciência, que antes tinha somente o papel investigativo e sociologicamente marginal, tornou-se hoje o centro da sociedade humana controlada pelos poderes econômicos, estatais e subjulgada a exigências dessa sociedade. Portanto, ao mesmo tempo que a ciência possui autoridade e é dominadora, ela também é dominada por essa demanda da sociedade meramente usuária. Isso é perigoso, vamos olhar como exemplo para um cenário particular de interesse aqui do blog; a biologia sintética é hoje uma área onde a sociedade está depositando muito de suas expectativas (e dinheiro). Esta pressão social por inovações pode ser bastante trágica nesta área, uma vez que pequenos bugs num produto biológico pode apresentar consequências drásticas.
O ponto principal a ser pensado aqui é a importância e necessidade que TODOS tenhamos consciência do que estamos criando e para que isto está sendo feito frente ao avanço cientifico e tecnológico. Para os envolvidos diretamente com ciência resta pensar no poder do que produzem, um poder pelo qual muitas vezes pode-se perder o poder. Instala-se a necessidade de uma verdadeira reflexão sobre os caminhos que os cientistas e o conhecimento científico podem seguir para que se tornem mais acessíveis. O avanço na divulgação científica tem que continuar crescendo, no entanto, prezando pela boa qualidade. Para os não envolvidos ansiosos pelas novidades do ano, pelos produtos recém-saídos do forno, embalados e “pronto para consumo” resta tomar uma postura crítica maior com relação a mudanças que afetam diretamente as suas vidas. Abrir caixas pretas é uma tarefa árdua mas o primeiro passo é se dar conta de que existem. Vale a tentativa(!!!) uma vez que o progresso da ciência e por sua vez de toda sociedade depende disso, depende das ideias inovadoras, das discussões, das críticas. Dependemos do conhecimento.
Autoria: Carolina Menezes Silvério e Marcelo Boareto.
Calculadora para sítios de ligação com ribossomos (RBS)
 Um objetivo central da biologia sintética é programar células para desenvolver funções valiosas. À medida que se constroem sistemas genéticas maiores e mais complexos (como os de escala genômica), serão necessários modelos e técnicas para combinar as partes genéticas de maneira eficiente para se atingir um comportamento específico. Para isso, serão necessários modelos biofísicos que descrevam a relação de uma sequência de DNA que a sua função. Um passo muito importante nesse sentido foi dado pelo Grupo do Prof. Howard Salis, pesquisador que eu tenho o prazer de trabalhar dentro do Synberc, com o desenvolvimento da calculadora de RBS (ribossomal binding site ou sítio de ligação com o ribossomo). Engenharia genética de microrganismos é um processo tempo intensivo (por ex. o desenvolvimento de uma nova rota metabólica para a produção de um produto químico pode levar de 5 a 10 anos de P&D para chegar a etapa industrial) que normalmente requer múltiplas rodadas de tentativas e erro utilizando mutações aleatórias. À medida que se torna possível construir sistemas gênicos cada vez mais complexo (incluindo genomas completos), métodos automatizados para montagem desses sistemas e para otimização de vias metabólicas se tornam necessários para diminuir custos e tempo de desenvolvimento. Além disso, com o aumento da complexidade do sistema, a aplicação de métodos de tentativa e erro para sua otimização se torna cada mais difícil e ineficaz. Uma maneira de otimizar um sistema gênico é através da variação da sequencia de seus elementos regulatórios para controlar os níveis de expressão de suas proteínas codificadoras. Cada passo limitante na expressão de um gene oferece a oportunidade para modular racionalmente os níveis de expressão proteica. Em bactérias, sítios de ligação do ribossomo e outras sequencias regulatórias de RNA são elementos de controle eficientes para o início da tradução. Como consequência, essas sequências são comumente modificadas para a otimização de circuitos genéticos. Vias metabólicas e expressão de proteínas recombinantes. Assita um video bem interessante no Youtube sobre tradução. Não é mostrado no video (e não consegui encontrar um melhor) o RBS é uma sequencia do RNA que direciona o ribossomo para o start codon, ele complementar a região do rRNA 16S que é parte da subunidade pequena 30S do ribossomo. Basicamente, quando mais complementar o RBS é ao 16S rRNA, maior é a afinidade e maior é a taxa de tradução. Como foi descrito no video, a tradução em bactérias (procariotos) consiste em quatro fases: iniciação, elongamento, terminação e o turnover do ribossomo (na verdade, esta última fase não foi mostrada no video). Na maioria dos casos, o início da transcrição é o gargalo do processo inteiro. O taxa de iniciação de transcrição se dá pela combinação de diferentes efeitos moleculares: incluindo a hibridação do rRNA 16S com a sequencia do RBS, a ligação do tRNA formilmetionina ao start codon, a distância entre o síto de ligação do rRNA 16S e o start códon, e a presença de estruturas secundárias de RNA que podem obstruir o RBS ou o start codon. Para o otimização de expressão de genes, é muito comum o desenvolvimento de bibliotecas de sequencias de RBS com o objetivo de otimização de funções de sistemas gênicos. Porém, a construção e seleção de bibliotecas de sequências se torna impraticável com o aumento de proteínas no sistema. Por exemplo, para realizar mutações randômicas em 4 nucleotídeos para um RBS resulta em uma biblioteca de 256 sequencias. O tamanho da biblioteca aumenta combinatoriamente com o número de proteínas do sistema, ou seja, 16,7 milhões de sequências para um sistema com 3 proteínas e 2,8 x 1014 sequencias para um sistemas com 6 proteínas). Dessa maneira, se torna necessários processos mais racionais para avaliar sequencias de RBS. A calculadora de RBS utiliza um modelo estatístico termodinâmico para predizer a taxa de iniciação de tradução de uma proteína. Dado um RBS e a região codificadora da proteína, o modelo é capaz de calcular a mudança de energia livre durante a montagem do complexo ribossomal 30S no RNAm (ΔGTOT). Depois, o modelo estatístico é capaz de correlacionar a taxa de início de transcrição com o ΔGTOT. Dessa maneira, o modelo biofísico preenche uma lacuna de desenho racional de RBS, criando uma relação quantitativa entre um sequencia de letras (As, Gs, Cs e Us) e um número (taxa de iniciação de tradução). A calculadora de RBS, portanto, combina um modelo biofísico com otimização estocástica para identificar uma sequência sintética (não natural) de RBS que irá proporcionar a taxa de início de tradução desejada. É importante destacar que esta relação também depende dos 35 nucleotídeos iniciais da região codificadora da proteína e que o RBS sintético precisa ser desenhada com esta sequencia incluída. A calculadora de RBS está disponível do site do laboratório do Salis . E é muito simples de utilizar, basta criar uma conta de usuário, recortar e colar as sequências, e definir uma ou mais taxas de iniciação de transcrição.
Um objetivo central da biologia sintética é programar células para desenvolver funções valiosas. À medida que se constroem sistemas genéticas maiores e mais complexos (como os de escala genômica), serão necessários modelos e técnicas para combinar as partes genéticas de maneira eficiente para se atingir um comportamento específico. Para isso, serão necessários modelos biofísicos que descrevam a relação de uma sequência de DNA que a sua função. Um passo muito importante nesse sentido foi dado pelo Grupo do Prof. Howard Salis, pesquisador que eu tenho o prazer de trabalhar dentro do Synberc, com o desenvolvimento da calculadora de RBS (ribossomal binding site ou sítio de ligação com o ribossomo). Engenharia genética de microrganismos é um processo tempo intensivo (por ex. o desenvolvimento de uma nova rota metabólica para a produção de um produto químico pode levar de 5 a 10 anos de P&D para chegar a etapa industrial) que normalmente requer múltiplas rodadas de tentativas e erro utilizando mutações aleatórias. À medida que se torna possível construir sistemas gênicos cada vez mais complexo (incluindo genomas completos), métodos automatizados para montagem desses sistemas e para otimização de vias metabólicas se tornam necessários para diminuir custos e tempo de desenvolvimento. Além disso, com o aumento da complexidade do sistema, a aplicação de métodos de tentativa e erro para sua otimização se torna cada mais difícil e ineficaz. Uma maneira de otimizar um sistema gênico é através da variação da sequencia de seus elementos regulatórios para controlar os níveis de expressão de suas proteínas codificadoras. Cada passo limitante na expressão de um gene oferece a oportunidade para modular racionalmente os níveis de expressão proteica. Em bactérias, sítios de ligação do ribossomo e outras sequencias regulatórias de RNA são elementos de controle eficientes para o início da tradução. Como consequência, essas sequências são comumente modificadas para a otimização de circuitos genéticos. Vias metabólicas e expressão de proteínas recombinantes. Assita um video bem interessante no Youtube sobre tradução. Não é mostrado no video (e não consegui encontrar um melhor) o RBS é uma sequencia do RNA que direciona o ribossomo para o start codon, ele complementar a região do rRNA 16S que é parte da subunidade pequena 30S do ribossomo. Basicamente, quando mais complementar o RBS é ao 16S rRNA, maior é a afinidade e maior é a taxa de tradução. Como foi descrito no video, a tradução em bactérias (procariotos) consiste em quatro fases: iniciação, elongamento, terminação e o turnover do ribossomo (na verdade, esta última fase não foi mostrada no video). Na maioria dos casos, o início da transcrição é o gargalo do processo inteiro. O taxa de iniciação de transcrição se dá pela combinação de diferentes efeitos moleculares: incluindo a hibridação do rRNA 16S com a sequencia do RBS, a ligação do tRNA formilmetionina ao start codon, a distância entre o síto de ligação do rRNA 16S e o start códon, e a presença de estruturas secundárias de RNA que podem obstruir o RBS ou o start codon. Para o otimização de expressão de genes, é muito comum o desenvolvimento de bibliotecas de sequencias de RBS com o objetivo de otimização de funções de sistemas gênicos. Porém, a construção e seleção de bibliotecas de sequências se torna impraticável com o aumento de proteínas no sistema. Por exemplo, para realizar mutações randômicas em 4 nucleotídeos para um RBS resulta em uma biblioteca de 256 sequencias. O tamanho da biblioteca aumenta combinatoriamente com o número de proteínas do sistema, ou seja, 16,7 milhões de sequências para um sistema com 3 proteínas e 2,8 x 1014 sequencias para um sistemas com 6 proteínas). Dessa maneira, se torna necessários processos mais racionais para avaliar sequencias de RBS. A calculadora de RBS utiliza um modelo estatístico termodinâmico para predizer a taxa de iniciação de tradução de uma proteína. Dado um RBS e a região codificadora da proteína, o modelo é capaz de calcular a mudança de energia livre durante a montagem do complexo ribossomal 30S no RNAm (ΔGTOT). Depois, o modelo estatístico é capaz de correlacionar a taxa de início de transcrição com o ΔGTOT. Dessa maneira, o modelo biofísico preenche uma lacuna de desenho racional de RBS, criando uma relação quantitativa entre um sequencia de letras (As, Gs, Cs e Us) e um número (taxa de iniciação de tradução). A calculadora de RBS, portanto, combina um modelo biofísico com otimização estocástica para identificar uma sequência sintética (não natural) de RBS que irá proporcionar a taxa de início de tradução desejada. É importante destacar que esta relação também depende dos 35 nucleotídeos iniciais da região codificadora da proteína e que o RBS sintético precisa ser desenhada com esta sequencia incluída. A calculadora de RBS está disponível do site do laboratório do Salis . E é muito simples de utilizar, basta criar uma conta de usuário, recortar e colar as sequências, e definir uma ou mais taxas de iniciação de transcrição. 
Outras ferramentas para controle de transcrição também estão disponíveis como a Small RNA Calculator.
Bons experimentos!
Salis, H., Mirsky, E., & Voigt, C. (2009). Automated design of synthetic ribosome binding sites to control protein expression Nature Biotechnology, 27 (10), 946-950 DOI: 10.1038/nbt.1568
Modelagem em biologia (sintética), um guia para ateus em modelagem.
 Para uma pessoa que lida diariamente com biologia, pode ser bastante difícil imaginar uma maneira de abordar matematicamente um problema em sua área ou como isto poderia ter alguma utilidade. A biologia é uma ciência considerada bastante complexa, pois são muitas as variáveis que podem afetar o sistema. Até mesmo um sistema relativamente simples como a expressão de uma proteína em E. coli, pode se tornar um problema bastante complexo de se modelar se todas as variáveis que afetam a expressão desta proteína forem levadas em consideração. Na realidade, não há nem mesmo quem seja capaz de listar todas estas variáveis. Assim sendo, como é possível fazer um modelo disto, se não consigo nem mesmo listar as variáveis que alteram meu sistema? Neste caso, melhor mesmo fazer como o Calvin e ser um ateu em modelagem, não é mesmo?
Para uma pessoa que lida diariamente com biologia, pode ser bastante difícil imaginar uma maneira de abordar matematicamente um problema em sua área ou como isto poderia ter alguma utilidade. A biologia é uma ciência considerada bastante complexa, pois são muitas as variáveis que podem afetar o sistema. Até mesmo um sistema relativamente simples como a expressão de uma proteína em E. coli, pode se tornar um problema bastante complexo de se modelar se todas as variáveis que afetam a expressão desta proteína forem levadas em consideração. Na realidade, não há nem mesmo quem seja capaz de listar todas estas variáveis. Assim sendo, como é possível fazer um modelo disto, se não consigo nem mesmo listar as variáveis que alteram meu sistema? Neste caso, melhor mesmo fazer como o Calvin e ser um ateu em modelagem, não é mesmo?
Na verdade não, fazer um modelo não é tão complicado assim. Pensar que é necessário colocar tudo no modelo é o erro conceitual mais comum de quem tem formação em áreas complexas como a biologia. Já os físicos por exemplo, tem uma visão dita reducionista, de tentar entender um problema dividindo-o em pequenas partes fundamentais e começar pelo modelo mais simples possível para depois aumentar a complexidade, se necessário (ou possível). Reza a lenda que um dono galinheiro certa vez chamou um fisico para solucionar o porquê as galinhas não estavam botando. Uma semana depois o fisico apareceu com a solução. Entretanto, a solução só era válida para galinhas esfericamente simétricas e no vácuo. True story!
Pode parecer contraditório, mas um modelo que leva mais variáveis em consideração não necessariamente é melhor ou mais “realistico”. Na verdade se um modelo leva mais variáveis em consideração do que outro e ambos tem a mesma efeciência, o segundo modelo é considerado melhor, conforme veremos.
Portanto, o melhor caminho para fazer um bom modelo é considerar as poucas e relevantes variáveis do problema, ou seja, ao fazer um modelo, a principal regra é:
Keep it simple, stupid!!
Este é o famoso princípio KISS, uma boa regra para começar um modelo. A maioria dos modelos funcionam melhor e são melhor entendidos se mantidos simples. Complexidade desnecessária deve ser evitada, mas obviamente, simplicidade demasiada não deve resultar em um modelo útil (como no caso das galinhas). Assim, uma boa maneira de se começar um modelo é pensar quais são as variáveis que devem ser realmente importantes para o problema. Tente formular seu modelo com o mínimo de variáveis e veja se seus resultados condizem com o esperado, ou com os experimentos. Se isto não ocorrer, é um sinal de que seu modelo ou é demasiadamente simples e você esqueceu alguma variável muito importante ou você pode ter feito hipóteses que não sejam válidas. Fazer hipóteses condizentes não é uma tarefa na simples e exige um conhecimento profundo do problema em questão.
O principio KISS é um conceito bastante semelhante à famosa navalha de Occam. Este princípio, introduzido por William Occam diz:
“Se em tudo o mais forem idênticas as várias explicações de um fenômeno, a mais simples é a melhor.”
Exemplo
Para exemplificar o que foi dito, vamos brincar de modelar com um simples exemplo que discutimos certa vez em nosso grupo.
O problema consiste em estimar a concentração de uma proteína (nosso caso era a Cre recombinase) dentro das bactérias E.coli, ou seja, quantas Cre-recombinases existem, em média, por bactéria? Esta inferência é base para estimar o PoPS (veja post anterior)
Este parece ser um problema simples mas pode se tornar bastante complicado de resolver caso o princípio KISS não seja utilizado. Quem é importante neste problema? Devo considerar a temperatura? Devo considerar a quantidade de alimento no meio?
Você pode pensar, e com razão, que a temperatura e a quantidade de alimento são importantes pois afetam a taxa de produção das proteínas. Entretanto estes são exemplos de variáveis que não precisam
ser levados em consideração pois, nos experimentos não trabalharemos com situações extremas de escassez de alimento nem de mudanças de temperatura e pequenas variações destas variáveis (fora de um regime extremo) não devem afetar significantemente a produção da proteína. Muitas são as variáveis que não afetam significamente o sistema e ter intuição disto é fundamental e repito, exige um bom entendimento do problema.
OK, mas por onde começar?
Bom, sabemos que para se produzir uma proteína primeiramente precisamos da produção do RNA mensageiro. A quantidade de mRNA certamente é uma variável relevante!!!
Portanto, vamos tentar criar uma equação sobre como o mRNA deve variar no tempo. A variação temporal de uma variável é representada matematicamente pela derivada da variável no tempo ![]()
Sabemos que nosso mRNA deve ser produzido pela leitura do DNA, feita pela DNA polimerase. OK, mas com que velocidade ela lê isto? Uma boa referencia, é o Bionumbers (tipo um google para dados biológicos)
Lá encontramos que nossa taxa de transcrição (Ktrans) é de, em média, 40 pares de base por segundo. Mas então precisamos saber qual o tamanho do RNA que gera nossa proteína. No caso da Cre é de 1032 pares de base (Nbp). Portanto, quantidade de proteína produzida por tempo e por volume (V) é de:
![]()
Dividimos pelo volume pois queremos saber a variação de concentração, ou seja, quantidade de proteínas por volume (unidade em Molar). Este será o primeiro termo de nossa equação, que se refere a produção do mRNA. Existem outras maneiras dele ser produzido? Se sim, novos termos devem ser adicionados. Neste caso, aparentemente esta é a unica forma dele ser produzido. Mas ele pode ser degradado, não é mesmo? Então precisamos de mais um termo, o de degradação. Novamente se formos até o bionumbers teremos a taxa de degradação (KdRNA) do mRNA. Este novo termo fará com que a taxa do mRNA diminua no tempo, e por este motivo ele é negativo. Portanto nossa equação fica:
![]()
Onde o termo positivo se refere a produção e o negativo se refere a degradação.
Agora vamos escrever uma equação para a tradução do mRNA em proteína. Neste caso encontramos uma taxa de tradução de 15 aminoacidos por segundo. Como nossa proteina tem 1032 pares de base ela deve ter 1032/3=344 aminoacidos. Como, além de produzida, nossa proteína também pode ser degradada então temos uma equação bastante semelhate à anterior:
![]()
Podemos supor que inicialmente a concentração desta proteína é zero, isto não fará diferença nos cálculos mas suponhamos que não haja proteina inicialmente. Ao longo do tempo, a concentração
desta proteína irá crescer até alcançar o equilibrio entre produção e degradação. Neste equilibrio, a concentração das proteínas não mudam mais no tempo e portanto nossas equações são iguais a zero. Para entender o equilibrio, pense na equação logistica que descreve a curva de crescimento de uma população de bactérias. Inicialmente temos um crescimento exponencial, mas depois de um tempo a população satura, ou seja, estabiliza em uma determinada população. Este ponto de saturação é que chamamos de ponto de equilibrio, onde a quantidade de bactérias não muda mais no tempo. Neste ponto, a quantidade de bactérias que morrem é proporcional às que “nascem”. Matematicamente o ponto de equilibrio é um ponto onde a derivada no tempo é igual a zero, portanto:
![]()
![]()
ou seja:
![]()
e
![]()
Agora podemos isolar a concentração do mRNA na primeira equação e substituir na segunda. Com isto, chegamos a:
![]()
Qual o sentido deste valor? Bem você pode utilizar o volume da bactéria e calcular qual a concentração de uma única proteína dentro de uma bactéria e você chegará que isto é aproximadamente 1 nM. Portanto, nosso resultado nos diz que há aproximadamente 2.000 proteínas, em média, dentro da bactéria.
OK, isto quer dizer que se eu fizer um experimento eu vou encontrar exatamente 2000 proteínas dentro da bactéria?
Obviamente não, devemos ter em mente a limitação de nosso modelo. Aproximações foram feitas e há muitas variáveis que podem fazer com que este valor mude. Entretanto, podemos dizer com certa segurança que teriamos algo de 1.000 à 10.000 proteínas na bactéria. Pode parecer muito inexato e que nosso modelo não foi tão útil por não ser preciso. Mas devemos lembrar que inicialmente não tínhamos nenhuma ideia de quantas proteínas haviam. Se alguém chutasse que há somente 10 ou 100 proteínas em média poderiamos pensar que era uma estimativa boa. Com o modelo sabemos que esta estimativa não é boa, que devem haver bem mais proteínas que isto!
Além da quantidade de proteína, com este simples modelo poderiamos estimar o tempo que demora para que a quantidade de proteína sature, ou seja, atinja o ponto de equilibrio. Estas são estimativas que podem ser muito úteis na hora de definir um protocolo experimental e pode economizar uma razoável quantidade de tempo e reagentes durante os experimentos. Portanto, não é necessário de ser ateu em modelagem, mas, tampouco, é recomendado acreditar religiosamente no modelo!!
Polimerase Por Segundo
![]() A Biologia é imprecisa por natureza, e vice versa. Isso é uma grande dificuldade ao se fazer design de sistemas biológicos sintéticos; aquilo que é muito bonito no papel às vezes nunca pode ser feito por motivos obscuros e por excesso de ruído dos sinais do sistema. Não dá pra prever. Na tentativa de deixar dispositivos sintéticos mais previsíveis, a Biologia Sintética tenta padronizar não somente partes biológicas, mas também os sinais que a compõem a dinâmica de seu sistema. Esses sinais são justamente a passagem de informação entre DNA e o fenótipo desejado, mas… como diabos deixar isso mais preciso e medir a velocidade dessa passagem de informação? Como medir “Polimerases Por Segundo”?
A Biologia é imprecisa por natureza, e vice versa. Isso é uma grande dificuldade ao se fazer design de sistemas biológicos sintéticos; aquilo que é muito bonito no papel às vezes nunca pode ser feito por motivos obscuros e por excesso de ruído dos sinais do sistema. Não dá pra prever. Na tentativa de deixar dispositivos sintéticos mais previsíveis, a Biologia Sintética tenta padronizar não somente partes biológicas, mas também os sinais que a compõem a dinâmica de seu sistema. Esses sinais são justamente a passagem de informação entre DNA e o fenótipo desejado, mas… como diabos deixar isso mais preciso e medir a velocidade dessa passagem de informação? Como medir “Polimerases Por Segundo”?
Padronização da Transmissão de Informação
Independente do que um aparelho elétrico faça, existem sinais “universais” que pertencem a todos eles: variações na diferença de potencial, na corrente, no campo elétrico e etc. A transmissão de informação entre os dispositivos eletrônicos que compõem esse aparelho são dadas justamente através desses sinais, fazendo todo o sistema elétrico funcionar. Em circuitos genéticos, sinais análogos à corrente elétrica são as taxas de transcrição e tradução, ou mais especificamente, a velocidade com que – respectivamente – uma polimerase e um ribossomo “leêm” seus nucleotídeos. O problema é que esses sinais (as taxas de transcrição e tradução) não são bons como transmissores de sinais. Entenda o porquê:
PoPS e RiPS: Qual é o sentido disso!?
Para que um transmissor de sinal seja bom, ele precisa facilitar com que dispositivos possam ser facilmente combinados em um sistema – além de ser algo “universal”, como foi dito anteriormente. Foi aí então que, usando experiências da engenharia, os biólogos sintéticos cunharam o termo “PoPS” (Polimerase Per Second – Polimerase Por Segundo) e “RiPS” (Ribossome Per Second – Ribossomo Por Segundo). Muitos pesquisadores acham que a criação desses novos termos é como “reinventar a roda”: qual seria a grande diferença entre isso e as clássicas taxas de transcrição e tradução? A diferença é a abrangência da nova medida. Quando se trata de um sítio operador, um RBS, um RNAm e o próprio gene sendo “lido”, não há diferença alguma em se medir uma taxa de transcrição e o “PoPS” ou uma taxa de tradução e o “RiPS”. Mas faz sentido se medir a taxa de transcrição de um sítio terminador por exemplo!? Esse elemento de DNA, que teoricamente não é transcrito (é ele quem justamente para a transcrição), ainda pode eventualmente ter um “leak” e permitir a passagem de uma polimerase. Usar a expressão “… a taxa de transcrição de um sítio terminador …” não faz sentido nenhum, mas acontece. Se usarmos PoPS, que por definição é o número de vezes que uma RNA polimerase passa por um ponto específico de uma molécula de DNA por unidade de tempo, ainda há sentido, pois nessa definição não importa qual a região do DNA a Polimerase passa. É esse tipo de generalidade que permite o fácil uso e novas combinações de dispositivos sintéticos.
Hierarquia de Abstração
Com a criação de sistemas fáceis de se integrar, os engenheiros biológicos podem se beneficiar de métodos largamente praticados em qualquer campo da engenharia, como a hierarquia de abstração. Com isso é mais simples se lidar com a complexidade de sistemas biológicos quando se omite informações desnecessárias. Desse modo (ver imagem abaixo), alguém trabalhando no nível de abstração das partes biológicas não precisa se preocupar com o design e síntese do DNA que usará, do mesmo modo, alguém trabalhando no nível sistêmico precisa pensar em apenas quais dispositivos incluir e como conectá-los para realizar uma função desejada, sem precisar se preocupar com os outros níveis de abstração.

Imagem retirada de: D. Baker, G. Church, J. Collins, D. Endy, J. Jacobson, J. Keasling, P. Modrich, C. Smolke, and R. Weiss. ENGINEERING LIFE: Building a FAB for biology. Scientific American, pages 44–51, June 2006.
Como medir PoPS?
A maioria dos sistemas criados e estudados hoje em dia em Biologia Sintética envolve controle transcricional da atividade genética, o que faz do PoPS a variável mais difundida na área, principalmente pelas pesquisas envolvendo lógica booleana em sistemas genéticos (portanto não é muito comum encontrar “RiPS” em artigos por aí).
Não existe um método direto para se medir PoPS, mas é possível chegar em seu valor indiretamente através de medições de fluorescência de genes reporter. É possível – se você puder encontrar os parâmetros na literatura ou medí-los – encontrar o PoPS de um dispositivo em cinco passos:
Cinco Passos Para o PoPS
1. Ligação de um Gene Repórter como Output
Antes de mais nada, será preciso de um fluorímetro (é claro) e demum espectofotômetro para medir densidade celular. Como exemplo, vamos observar a parte BBa_F2620:

Esse BioBrick tem como “entrada” a substância de quorum sensing 3-oxohexanoil-homoserina lactona e tem como “saída” PoPS. Em presença de 3OC6HSL, o gene que produz o fator de transcrição luxR promove a transcrição de genes após o Lux pR, na parte final do BioBrick BBa_F2620. Para mensurar o quão ativo o luxpR fica, liga-se outro BioBrick no final do dispositivo para mudar o output do sistema colocando-se o BBa_E0240 – a ORF (Open Reading Frame) do GFP (Green Fluorescent Protein):

Assim tem-se uma nova parte, o BioBrick BBa_T9002:

2. Medição da Fluorescência e Absorbância e Subtração do Background
Para medir a fluorescência do GFP e a absorbância da amostra de células, é preciso criar dois controles: um da absorbância (A) e outro da fluorescência (G). O controle da fluorescência será o próprio BBa_T9002 sem ser induzido pela substância de quorum sensing (G_não-induzido), enquanto o controle da absorbância é feita da maneira trivial, verificando somente a absorbância do meio de cultura (A_background). Para se obter os reais valores de Fluorescência induzida por 3OC6HSL e da densidade celular, basta então subtrair esses valores de background com os valores medidos durante a indução pela substância de quorum sensing:

3. Correlação com a Curva Padrão
Com as correções em mãos, outro procedimento trivial a ser feito é encontrar a curva padrão de fluorescência versus GFP e de absorbância versus número de células. Por exemplo, experimentos feitos em laboratório chegaram a essas retas de correlação de valores:

Em que UFC é “Unidade Formadora de Colônia” – o número de células na amostra. E “GFP” seria o número de moléculas de GFP medidas.
4. Interpolar a Curva de GFP versus Tempo Obtida na Medição
A síntese total de GFP por célula (S_célula) é dada pela taxa de produção de GFP total (S_total) dividida pelo número de células (UFC):

Para encontrar a derivada de [GFP] por tempo, basta plotar os dados de GFP obtidos por tempo e interpolar com uma função logística (provavelmente) para obter a equação que melhor descreve a variação de GFP no tempo.
5. Colocar os Valores Nessa Equação Aqui
Depois de determinada a função Scélula, basta colocá-la nessa fórmula e encontrar o PoPS:

Em que:
a = Taxa de maturação do GFP – 1/s
GammaM = Constante de degradação do RNA – 1/s
GammaI = Constante de degradação do GFP imaturo – 1/s
Rô = Constante de síntese proteica por RNAm (RiPS) – [Proteína]/[RNAm].s
PoPS = Polimerase por segundo – [mRNA]/[DNA].s
CUIDADO: Conteúdo Matemático – Prossiga com Cuidado (Ou não…)
Chega-se nessa expressão através de um pequeno sistema de equações diferenciais:

As equações expressam uma dinâmica simplificada de um sistema de transcrição e tradução de uma informação genética. Para chegar na expressão de PoPS, basta substituir a última equação na segunda e isolar M. Com a expressão resultante, basta substituir a variável M na primeira equação e sua derivada em dM/dt.
ATENÇÃO: Aqui acaba o conteúdo matemático. Está tudo bem agora.
E essa é a história de como você pode encontrar o PoPS – essa variável estranha! – no seu próprio laboratório (ou ao menos entender do que se trata). Assim como um circuito elétrico, que pode ser montado da melhor maneira possível e mesmo assim não funcionar por razões obscuras, sistemas biológicos têm muito mais esse problemático costume de não se comportar como esperado. Contudo essa abordagem mais generalista da atividade transcricional de uma célula é uma boa maneira de se tentar enfrentar o grande desafio de se deixar a biologia mais “engenheirável” e mais “precisa”. Não que essa seja a coisa mais fácil do mundo, mas ela nunca será se ninguém tentar. E estamos aos poucos conseguindo.
Referências:
- Bio FAB Group, Baker D, Church G, Collins J, Endy D, Jacobson J, Keasling J, Modrich P, Smolke C, & Weiss R (2006). Engineering life: building a fab for biology. Scientific American, 294 (6), 44-51 PMID: 16711359
- http://partsregistry.org/Part:BBa_F2620:Experience/Endy/Data_analysis
- http://openwetware.org/wiki/Endy:Measuring_PoPS_on_a_plate_reader
Entenda a Engenharia Metabólica
![]() Uma das grandes maravilhas da humanidade – objeto de grande satisfação entre os químicos – é uma tabela que nos diz tudo o que existe no universo, os cerca de 120 elementos que formam tudo aquilo que o ser humano conseguiu perceber. Usando essa mesma ideia, cientistas conseguiram determinar 12 substâncias principais que podem produzir tudo… o que existe dentro de uma célula! Esse é um dos princípios fundamentais da Engenharia Metabólica, entenda o porquê:
Uma das grandes maravilhas da humanidade – objeto de grande satisfação entre os químicos – é uma tabela que nos diz tudo o que existe no universo, os cerca de 120 elementos que formam tudo aquilo que o ser humano conseguiu perceber. Usando essa mesma ideia, cientistas conseguiram determinar 12 substâncias principais que podem produzir tudo… o que existe dentro de uma célula! Esse é um dos princípios fundamentais da Engenharia Metabólica, entenda o porquê:
Os 12 Precursores Principais
Tudo o que uma célula consome sempre produz compostos que chamamos de “precursores principais”. São esses precursores que podem gerar tudo dentro da célula: desde seu DNA até às membranas celulares. Na bactéria E.coli, por exemplo, existem 12 dessas substâncias principais: Eritrose 4-fosfato, o famoso Acetil CoA, Frutose 6-fosfato, Glucose 6-fosfato, Alfa-cetoglutarato, Oxaloacetato, Ribose 5-fosfato, Fosfoenolpiruvato, 3-fosfoglicerato, Piruvato (esse carinha é famoso também), Triose-fosfato e Succinil CoA. Isso quer dizer que a grande maioria de todas a milhares de reações dentro da E.coli em algum momento formam e/ou consomem essas substâncias em suas etapas de reação.

Assim, ao melhor estilo dos antigos alquimistas, pesquisadores – em especial FC Neidhardt – dissecaram células de E.coli de modo a determinar a quantidade desses precursores que seria necessária para “construir” uma bactéria (ver infográfico acima):

Ou seja, todos os precusores somados às moléculas para se realizar oxidações (NAD), reduções (NADPH) e fornecer energia (ATP), resultam em 1 mol de “XR”, que é a quantidade de biomassa produzida com esses compostos, ou “1 mol de células” (definida aqui como a quantidade de células em 10^6g). XR seria um arcabouço que abarca todas as proteínas, lipídeos e nucleotídeos da célula; por isso não podemos dizer que essa é de fato uma equação química, mas uma “pseudo-equação química”, afinal dá pra ver claramente que as quantidades das substâncias não se conservam em termos estequiométricos – pra falar a verdade, não há nem a representação de elementos, são só siglas.
Enfim, esse é o mais próximo que chegamos do desejo dos alquimistas de obter uma receita para a vida como eles idealizaram, mas apesar de parecer pouco, essa pseudo-reação global de “construção de células” nos permite contabilizar literalmente quais são os recursos que as bactérias têm para produzir coisas que não produzem natualmente, ou seja, nos mostram quais são as cartas em jogo quando se altera um organismo geneticamente. E o nome desse jogo é fluxo, fluxo metabólico.

O Fluxoma
Uma célula é como se fosse uma mini indústria: seus operários são enzimas, a chefia é a informação genética e a matéria prima são os metabólitos externos com o qual se produzem as peças – que são os 12 metabólitos principais – para a linha de montagem: as etapas de reações bioquímicas. Essa pequena empresa é um empreendimento talhado pelo mercado competitivo, ditado pela economia minimizadora de enegia, seguindo a lógica da seleção natural. Igualzinho às empresas de verdade. Mas enfim, a grande pergunta é: o que acontece quando a chefia muda? O que acontece quando modificamos geneticamente um microrganismo? Apenas colocar uma informação genética não natural na “chefia” é o mesmo que colocar um administrador inexperiente no comando de todo um processo produtivo que ele não conhece. É ir contra milhares de anos de seleção natural.

Arte de Pedro Pantai. Visite http://meninodacaixadesapato.blogspot.com.br/
Por exemplo, imagine que a nossa célula é uma fábrica de motos. Depois de muitos anos existindo, decidem colocar uma nova chefia adjunta no comando. O novo chefe adjunto decide colocar uma nova maquinaria e funcionários no chão de fábrica, pois quer ampliar a gama de produtos que a empresa fabrica. A indústria de motos então passa a produzir triciclos; nada mal. O problema é que a nova chefia SÓ faz isso. Ele não comunica os antigos funcionários sobre a nova produção, não compra mais matéria prima e, apesar de desejar que o carro chefe da empresa seja triciclos, não move uma palha para que isso aconteça. Em outras palavras: temos uma fábrica de motos que improvisa na fabricação de triciclos. É aí que entra o engenheiro de produç… Ops, o “engenheiro metabólico”.
O grande problema da nossa indústria de motos é apenas de distribuição das peças, afinal – simplificadamente – a grande diferença do produto antigo para o novo é apenas uma roda. Da mesma maneira, em uma célula a grande diferença entre os componentes que ela já produz para existir (o “XR” da pseudo-reação acima) e as novas substâncias que queremos que ela produza (por modificações genéticas) é apenas uma combinação de quantidades diferentes dos 12 precursores principais que levem às reações de síntese que queremos. Para ter controle dessas reações que levam à XR e/ou ao bioproduto desejado, cria-se o chamado “fluxoma”, a contabilização de todos as taxas de reação (os fluxos) de dentro da célula – da mesma forma que o genoma é a contabilização de toda a informação genética de uma célula.
ATENÇÃO: se a matemática não é sua amiga, tome cuidado com o conteúdo a seguir.
Fluxos Metabólicos
A teoria que se aplica para a determinação desses fluxos baseia-se na simples conservação de masa em um sistema fechado, no caso uma célula ou um compartimento celular fechado com metabólitos; especificando a reversibilidade das reações e quais metabólitos são considerados como internos e externos. A equação geral que descreve a conservação de massa de metabólitos em um sisema de volume definido pode ser escrita como:

Em que C (mol/L) é um vetor da concentração de m metabólitos internos; r ((mol/L)/h) é o vetor do grau de reação, ou seja o fluxo, de n reações que convertem metabólitos; S é a matriz estequiométrica de dimensões m x n cujos elementos sij representam o coeficiente estequiométrico do elemento i envolvido na reação j; e μ (1/h) é o grau específico de diluição associado com a mudança no volume de um sistema, o que é muito importante considerar no modelo, pois o graus de diluição afetam diretamente as velocidades de reação. Como em uma célula o grau de diluição é muito baixo quando comparado com os graus de reação, as mudanças de volume no sistema são consideradas negligenciáveis. Temos portanto a equação mais simplificada:

Em um estado estacionário, que é o que se considera na análise de um fluxo metabólico, não há acúmulo de metabólitos, e portanto suas concentrações, bem como a população bacteriana, tornam-se constantes, fazendo com que dC/dt = 0:
![]()
A caracterização de reações reversíveis é realizada através da detreminação do sinal de ri, em que ri < 0 delimita a reação ocorrendo no sentido oposto, ri = 0 informa a sua não-ocorrência e ri > 0 indica uma reação ocorrendo no sentido esperado.
Uma outra maneira mais simplista de se entender o mesmo raciocínio, partindo do mesmo princípio de conservação de massa, pode ser:

O que é o mesmo que S.r = 0. Considerando as substâncias envolvidas em várias reações, teremos o mesmo resultado:

OBSERVAÇÃO: Aqui acaba o conteúdo matemático. Pode continuar a ler abaixo, já passou…!
Análise de Vias Metabólicas
Então, como dá pra perceber, tudo se resume a encontrar um sistema de equações – sim, os sisteminhas de equações que você aprende a resolver na escola – que descreva o metabolismo da célula envolvendo os metabólitos principais. É exatamente aqui que entram os dados da pseudo-reação global comentada no início, é ela que define, junto com dados experimentais de consumo de substratos, o conjunto de soluções desse sistema de equações (chamados de “modos elementares”). Os sistemas de equações obtidos por análise das vias metabólicas são sempre indeterminados, uma vez que o número de reações bioquímicas as envolvendo é muito maior que o número de espécies de metabólitos, ou seja: tem-se mais equações que variáveis. A tarefa de programas de análise de vias metabólicas é encontrar possíveis soluções para esse sistema que digam quais são os possíveis fluxos de todas as reações envolvidas, com isso é possível analisar qual modo elementar é o que possui maior rendimento de produção do bioproduto desejado, e portanto quais reações que devem ocorrer no sistema em detrimento de outras.
Por exemplo, vejamos o exemplo da produção de Lisina em Corynebacterium glutamicum. Esse aminoácido é naturalmente produzido em nível basal na célula para manutenção da atividade celular, apenas super-expressando os genes envolvidos nas vias de produção de lisina e nocauteando outros genes que produzem enzimas competidoras (essas são grandes maneiras de se alterar os fluxos metabólicos) da biosíntese de lisina é possível aumentar cerca de 11 vezes a produtividade. Isso pode ser feito sem análise nenhuma. Mas se analisando os fluxos metabólicos (imagem abaixo), vemos que é possível quase dobrar a produção industrial de Lisina à partir da mesma quantidade de glicose. Assim como na analogia entre a indústria e a célula, única diferença foi a distribuição dos fluxos entre os precursores principais da C. Glutamicum, ou seja uma combinação diferente de quantidades dos precursores em diferentes reações.

No caso, um aumento do fluxo metabólico pela via das pentoses (formando Ribulose 5-fosfato) em um processo sem produção de CO2 – realizando o ciclo do glioxilato – aumenta a produção por gerar mais NADPH, necessário na biosíntese de Lisina, e que não é produzida na via “normal” de degradação da glicose (via de Embden-Meyerhoff-Parnas).
O Futuro da Engenharia Metabólica
Muitos dizem que a engenharia metabólica será tão eficiente em otimizar os processos biotecnológicos que substituirá completamente os processos químicos orgânicos no futuro, afinal esse é o grande entrave para termos toda uma indústria baseada em uma bioprodução: os processos químicos são muito mais eficientes. Ter toda a indústria química baseada na produção de materiais por organismos nos daria um mundo mais ecológico e renovável. O grande passo para isso já foi dado com a “synthia“, a bactéria sintética de Craig Venter e seu grupo. O desafio de se fazer engenharia metabólica é justamente o problema que foi eliminado – EDITED: OK, não eliminado, mas amenizado – quando se nocauteou todos os genes não essenciais para a sobrevivência na bactéria produzida por Venter, pois qualquer nova via colocada no microrganismo já estaria quase completamente otimizada, uma vez que não existiriam fluxos “não essenciais” em que a bactéria poderia estar “desperdiçando” energia em vez produzir o bioproduto dos genes com que foi modificada. Assim, como um upgrade da engenharia genética, a engenharia metabólica faz aquilo que torna a Biologia Sintética algo simples e bonito: apenas uma mudança inteligente de como a informação é transmitida; uma mudança de design. No final das contas, mais do que pseudo-realizar os sonhos dos alquimistas, entender os fluxos metabólicos é mudar a maneira como os químicos atuais sonham com o futuro, afinal, porque reinventar como produzir substâncias orgânicas se os próprios organismos podem fazer isso pela gente!? Já está mais do que na hora de reinventarmos nossa indústria.
Referências
Vallino JJ, & Stephanopoulos G (2000). Metabolic flux distributions in Corynebacterium glutamicum during growth and lysine overproduction. Reprinted from Biotechnology and Bioengineering, Vol. 41, Pp 633-646 (1993). Biotechnology and bioengineering, 67 (6), 872-85 PMID: 10699864
Neidhardt, F. C., J. Ingraham, and M. Schaechter. 1990. Physiology of the Bacterial Cell: A Molecular Approach. Sinauer Associates, Sunderland, MA.